















Abstract
Background. Accumulating evidence has supported the effect of antibody-dependent cellular phagocytosis (ADCP) on the tumor microenvironment (TME) and cancer therapy. However, an ADCP-based signature to predict the prognosis of gastric cancer (GC) has not been established.
Objectives. We aimed to develop an ADCP-based signature to improve the prognosis prediction of GC.
Materials and methods. Antibody-dependent cellular phagocytosis genes that exhibited a differential expression were characterized, followed by the construction and validation of the ADCP-based signature. The potential association between the ADCP-based signature and TME was explored, and the features of the signature genes were investigated. Finally, a predictive nomogram was established based on the ADCP-based signature.
Results. Four ADCP-related genes, MKNK2, VCAN, LRAT, and GNGB, were identified to construct the ADCP-based signature, and a high ADCP score predicted an unfavorable prognosis in GC patients (p < 0.05). The ADCP-based signature was significantly associated with immune cells, immune checkpoints and immune signaling pathways (p < 0.05). Gastric cancer patients with high ADCP scores benefited less from immunotherapy compared to those with low ADCP scores. A nomogram including age, stage and risk score of the ADCP-based signature was constructed to predict the 1-, 3- and 5-year survival probabilities, with an area under the curve (AUC) of 0.669, 0.675 and 0.685, respectively.
Conclusions. The ADCP-based signature may serve as a new option for prognosis prediction and the personalized treatment of GC patients.
Key words: bioinformatics analysis, gastric cancer, tumor microenvironment, prognostic signature, antibody-dependent cellular phagocytosis
Background
Gastric cancer (GC) was the 5th most diagnosed cancer and the 4th most common cause of cancer death in 2020.1 The incidence and mortality of GC have been reduced in recent years as a result of the prevention and treatment of Helicobacter pylori and Epstein–Barr virus (EBV) infections.2, 3 However, the prognosis of GC patients continues to be unsatisfactory due to the impact of locally advanced and distant metastases.4, 5
Immunotherapy is a promising treatment strategy, but only a fraction of GC patients benefit from it.6 Also, the immunosuppressive microenvironment of tumors severely reduces the effectiveness of immunotherapy. Therefore, there is a strong need for precise immunotherapy and accurate efficacy prediction using immune-based biomarkers.
Antibody-dependent cellular phagocytosis (ADCP) is the mechanism that leads to the internalization and degradation of target cells through the activation of Fcγ receptors on the surface of macrophages to induce phagocytosis.7 It has been shown that the ADCP process can influence the evolution of the tumor microenvironment (TME). A previous study has found that rituximab results in the upregulation of multiple Fcγ receptors on macrophages, which correlates with their phagocytic response.8 In addition, anti-KIT antibodies have been observed to inhibit the growth of gastrointestinal stromal tumors by inducing the phagocytosis of macrophages.9 These studies suggest that ADCP may regulate the progression of different cancers. Hence, it is valuable to examine the role of ADCP-related genes in the progression of GC, as well as establish a relevant prognostic model for the treatment of GC.
In the present research, we conducted bioinformatics approaches to construct and validate an ADCP-based prognostic signature by employing The Cancer Genome Atlas (TCGA) database and the Gene Expression Omnibus (GEO) database. We further explored the role of the ADCP-based signature in the immune microenvironment. Our study can effectively predict the prognosis of GC patients and may provide new perspectives for the treatment of GC.
Objectives
We aimed to develop a robust ADCP-based signature to improve the predicted prognosis of GC.
Methods
Data collection and processing
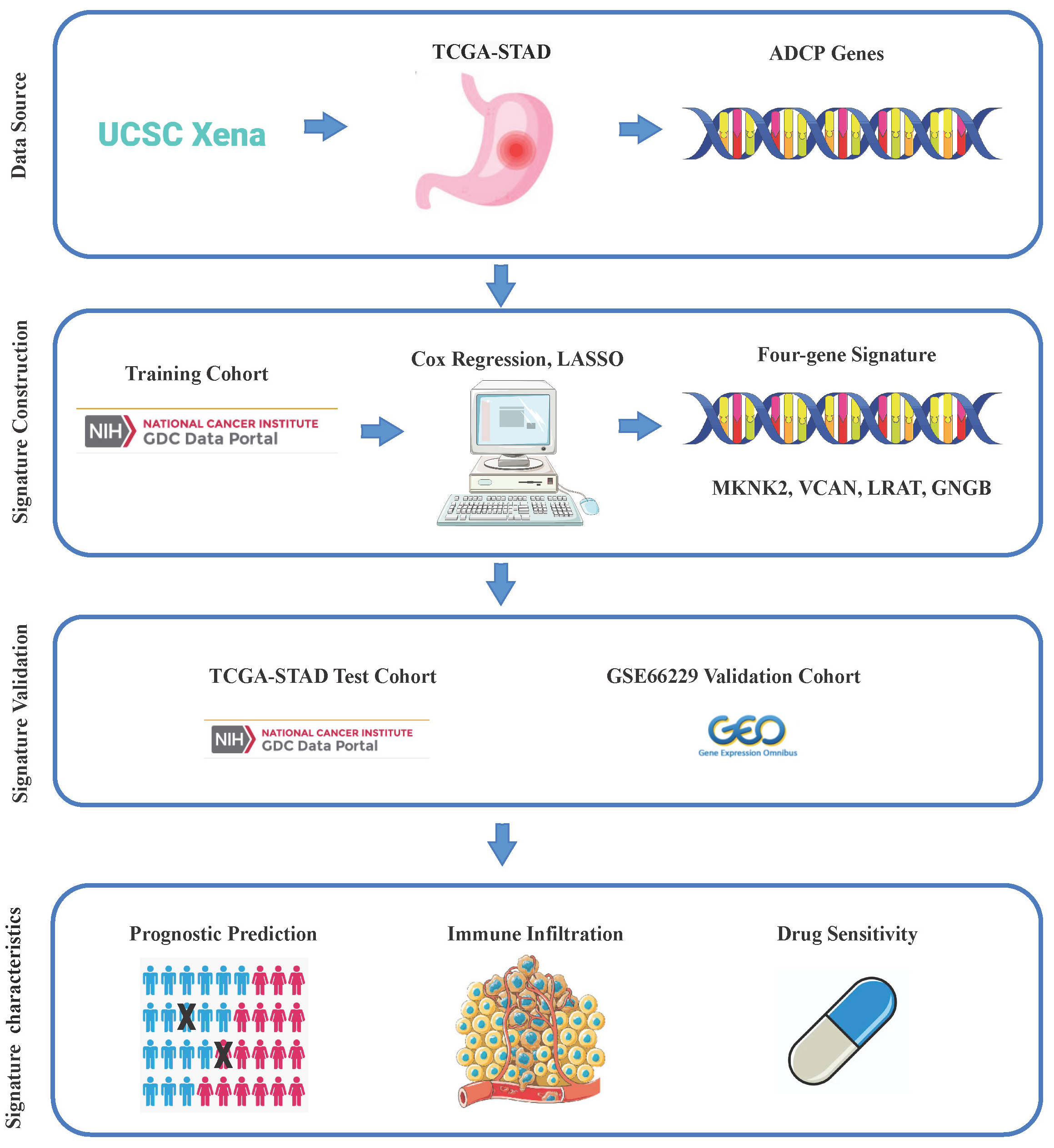
RNA-seq data and clinical information on TCGA-STAD were downloaded from the UCSC-Xena platform (https://toil.xenahubs.net), which contained 32 normal tissue samples and 375 tumor tissue samples. The dataset GSE66229 with 300 tumor tissue samples was downloaded from the GEO database for subsequent model validation analysis. A total of 3,405 genes were obtained by downloading ADCP-related genes (Supplementary Table 1) from a study by Kamber et al.10 and matching them with the above expression profiles. Our workflow is presented in Figure 1.
Protein–protein interaction network and functional enrichment analysis
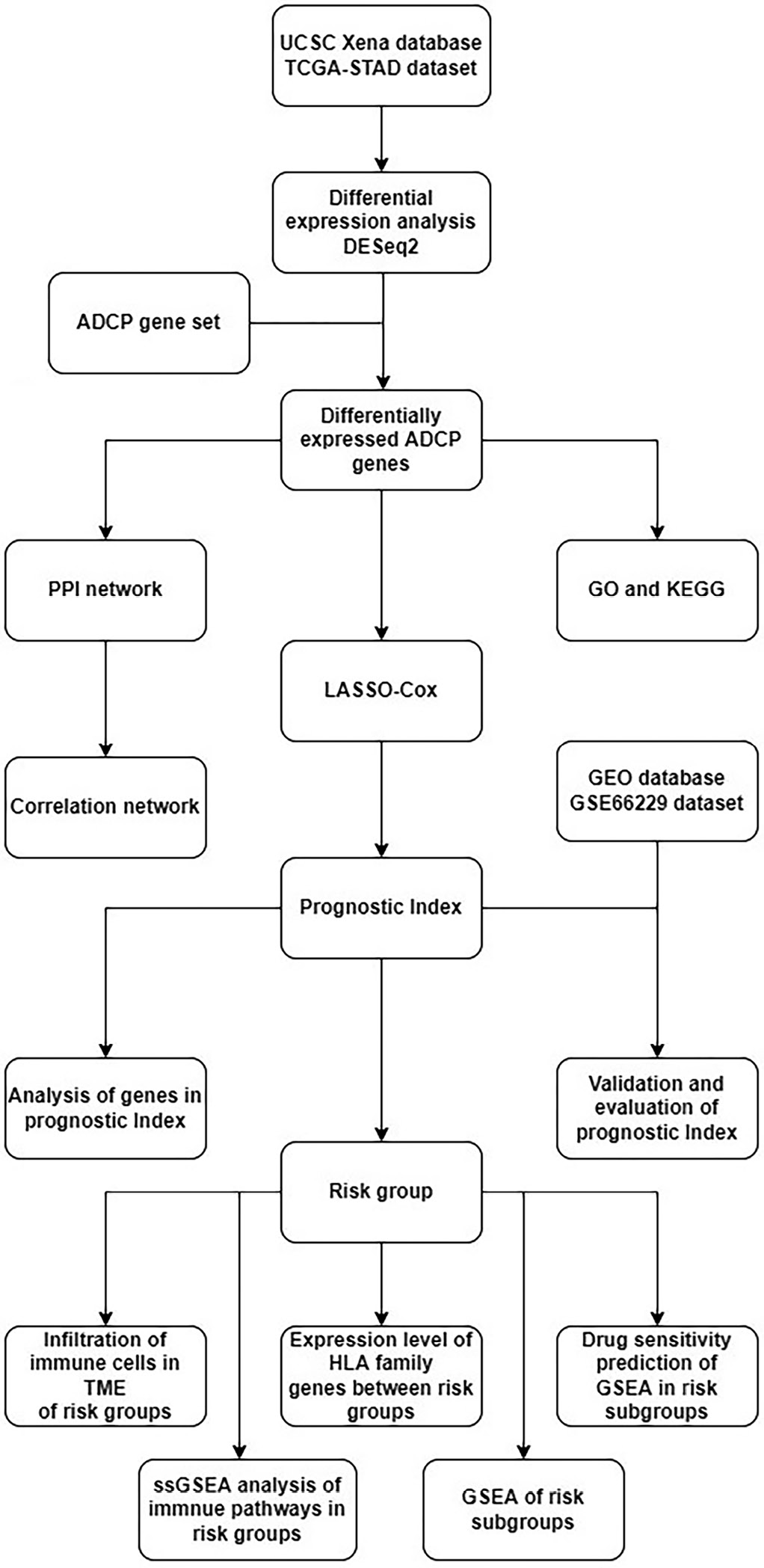
The differential expression analysis of ADCP genes was carried out using the R package DESeq2 (v. 1.36.0, https://www.bioconductor.org/packages/release/bioc/html/DESeq2.html). A false discovery rate (FDR) less than 0.05 and a |log2FoldChange| >1 were selected as the threshold for screening differentially expressed genes. The interaction relationship of the differentially expressed genes was analyzed using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (https://cn.string-db.org/) and imported into Cytoscape (v. 3.9.1) to map the protein-protein interaction (PPI) network. Gene Ontology (GO; http://www.geneontology.org) and Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.ad.jp/kegg) functional enrichment analysis was conducted using the ClusterProfiler package (v. 4.4.4, https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html).11
Construction and verification
of the ADCP-based signature
The 2 datasets from TCGA and GEO were log2-transformed and normalized to obey the same distribution, thus eliminating the effect from different batches. Tumor samples from TCGA-STAD were segregated into a training set and a test set with a 6:4 random split. Samples with survival times less than 30 days were filtered, and a total of 342 tumor tissue samples were finally included. In the training set samples, the differentially expressed genes and the survival information of the samples were merged to identify genes strongly related to the overall prognosis for survival using univariate Cox regression analysis performed with the survival package (v. 3.4-0, https://github.com/therneau/survival).12 The genes were further screened using a 10-fold cross-validation analysis using the executed least absolute shrinkage and selection operator (LASSO) Cox regression model employing the glmnet package (v. 2.0-18, httRiskscore://cran.r-project.org/web/packages/glmnet/index.html).13 Next, multivariable regression was conducted to further filter the genes. Relying on the gene regression prognostic coefficients and the expression levels of genes in the training set samples, a risk score model was developed; it was derived as follows:
risk score = Σ (Expi × Coefi);
Expi and Coefi represent the expression levels and LASSO regression coefficients of prognostic genes.
To inspect the correctness of the model, the risk score of each sample from the test set of TCGA and the external validation set of GEO was calculated using the same regression coefficients according to the risk score calculation formula. The samples from the 2 aforementioned validation sets were differentiated based on the median risk score as the cutoff. The overall survival (OS) of each group was assessed using Kaplan–Meier curves. We then plotted the 1-, 3- and 5-year receiver operating characteristic (ROC) curves using the R package survival ROC (v. 1.0.3, https://cran.rstudio.com/web/packages/survivalROC/index.html) and calculated the 1-, 3- and 5-year area under the curve (AUC) values, respectively.
Prognostic characterization of genes in the model and construction of ceRNA networks
We used the TCGA-STAD dataset for survival analysis of the ADCP-related genes in the signature. The multiMiR package was used to predict gene-associated microRNAs (miRNAs) in the model. Starbase was then used to predict lncRNAs that interact with miRNAs, and the CHEA3 database (https://maayanlab.cloud/chea3) was used to predict transcription factors (TFs) of the model genes. All results were imported into Cytoscape (https://cytoscape.org) to construct a competitive endogenous RNA (ceRNA) network.
Characteristics of the TME and GSEA in different subgroups
To further examine the immune microenvironment in high- and low-risk groups, Estimation of STromal and Immune cells in MAlignant Tumours using Expression data (ESTIMATE),14 Cell-type Identification by Estimating Relative Subsets of RNA Transcripts (CIBERSORT),15as well as single-sample gene set enrichment analysis (ssGSEA) algorithms were utilized to obtain TME scores and immune cell scores. Moreover, we extracted immune gene-related pathways through the immport database (https://www.immport.org/home). To assess immune pathway variations between high- and low-risk groups, the ssGSEA algorithm was used to calculate the immune pathway scores of cancer samples. We also captured expression data of human leukocyte antigen (HLA) family genes and immune checkpoint-associated genes to analyze their differential expression in high- and low-risk groups. Gene Set Enrichment Analysis (GSEA) was performed using the ClusterProfiler package (v. 3.18.0)11 to observe significant pathways enriched in the high- and low-risk groups.
Drug sensitivity and immunotherapy efficacy prediction
The sensitivity of patients to chemotherapeutic agents was evaluated using Genomics of Drug Sensitivity in Cancer (GDSC; https://www.cancerrxgene.org).16 The R software package pRRophetic (v. 0.5, https://github.com/paulgeeleher/
pRRophetic) was used to determine half-maximal inhibitory concentrations (IC50). We evaluated the differential in drug sensitivity between high- and low-risk groups. The Tumor Immune Dysfunction and Exclusion (TIDE) method was used to predict the benefit of immune checkpoint inhibitor (ICI) treatment in patients with GC.
Construction of nomogram
Clinical risk models were constructed using univariate and multivariate Cox regression analyses. Next, clinical risk values were calculated for all samples of TCGA-STAD. The prognosis was assessed using Kaplan–Meier curve analysis, and to test the clinical risk model, ROC curves were generated. The calibration curve analysis of the model was assessed using the R package rms (v. 6.3-0, https://hbiostat.org/r/rms). Using the R package dcurves (v. 0.3.0, https://github.com/ddsjoberg/dcurves), decision curve analysis (DCA) decision curves were produced to evaluate the clinical risk model.
Statistical analyses
All analyses were conducted in R v. 4.2.1 (R Foundation for Statistical Computing, Vienna, Austria), and a p-value of 0.05 or less was deemed statistically significant. Univariate and multivariate Cox regression analyses were performed to identify genes and clinical parameters significantly associated with the OS prognosis. Schoenfeld residual plots (conducted by the ggcoxzph function of the survival R package) were used to assess the proportional hazards assumption, which determines whether the effect of the variable on the hazard function is constant over time. The log-rank test was used to compare the differences in survival distributions. A Wilcoxon test was used to compare the differences in medians between samples. The key R-script used in this study can be found in the Supplementary materials.
Results
Differentially expressed ADCP genes and their function in GC
The TCGA dataset was used to obtain the differential expression matrix of GC, which was intersected with ADCP-related genes to obtain 531 differentially expressed ADCP-related genes (238 of them were upregulated and 293 were downregulated, Figure 2A, Supplementary Table 2). The most significantly up- and downregulated genes were plotted as a heat map (Figure 2B), with a total of 20 included. These 531 differentially expressed ADCP-related genes were mapped into a PPI network (Figure 2C, Supplementary Table 7) through the STRING database. Gene Ontology (Figure 2D, Supplementary Table 3) and KEGG analysis (Figure 2E, Supplementary Table 4) were used to explore the functions of the 531 genes. Functional investigation of differentially expressed ADCP-related genes revealed that they were involved in the p53 signaling pathway, PI3K/Akt signaling pathway, calcium signaling pathway, and platinum drug resistance (Figure 2E), which suggested the possible role of ADCP genes in cancer progression and metastasis.
Development and validation
of ADCP-based signature
Based on the above 531 differential genes, univariate Cox regression was applied using the TCGA-STAD training set to detect 7 prognosis genes (ADAMTS12, MKNK2, VCAN, MMP1, CLDN9, LRAT, and GNG8; Figure 3A, Supplementary Table 5). The results of the proportional hazards assumption can be found in Supplementary Fig. 1. To further screen the prognostic markers of GC, LASSO regression was employed to identify the prognostic genes acquired from the above univariate Cox regression. The optimal λ was obtained when the partial likelihood of deviance reached the minimum value (Figure 3B,C). Four prognosis-associated genes (MKNK2, VCAN, LRAT, and GNGB; Figure 3D, Supplementary Table 6) were available to construct ADCP-related gene signatures after performing multivariate Cox regression. Based on the median risk score, the samples from the training set were separated into a high-risk group (risk score >median risk score) and a low-risk group (risk score ≤median risk score). The ROC curve was plotted to display the 1-, 3- and 5-year AUC for the training set. Validation of the model was conducted in the TCGA test set and the GEO external validation set, indicating a good predictive performance of the model (Figure 3E). The prognosis differed significantly between the 2 groups, with patients in the high-risk score group presenting a poorer prognosis. In the TCGA-STAD training cohort of 206 GC patients, those with high-risk scores (50%) had a shorter OS (p = 5e-07) than those with low-risk scores (50%). High-risk patients (52.941%) had a shorter OS than low-risk patients (47.059%) across all 136 GC patients in the TCGA-STAD test cohorts (p = 0.003; Figure 3F). To identify whether or not the ADCP-based signature was reliable, the accuracy of the ADCP-based signature was evaluated using the GEO external validation cohort, with a total of 300 GC patients participating in this study. As seen in Figure 3F, high-risk patients (49%) had a shorter OS than the low-risk group (51%) (p = 5e-05). The prognostic information of the 4 genes in the signature was demonstrated using a Kaplan–Meier curve, while 3 genes showed significant prognostic value in GC. Between the MKNK2 subgroups, no OS differences were observed (log-rank test, p = 0.151, Figure 4A). In the VCAN high-expression group, GC patients were found to have a lower survival probability (log-rank test, p = 0.027, Figure 4B). Also, in the LRAT high-expression group, GC patients had a significantly lower OS (log-rank test, p = 0.01, Figure 4C). The high GNG8 expression group indicated a more favorable prognosis (log-rank test, p = 0.011, Figure 4D). Based on the 4 genes, 61 miRNAs were predicted using multiR, and 52 lncRNAs were further predicted with starbase (http://starbase.sysu.edu.cn), followed by the TFs of genes predicted with the CHEA3 database, and the top 10 TFs of meanRank were selected. Genes, miRNAs, lncRNAs, TFs, and interactions between them were imported into Cytoscape to map the ceRNA-TF network.
ADCP-based signature in TME
To gain further insight into the involvement of ADCP-based signature in TME, we used the CIBERSORT (Figure 5A), ssGSEA (Figure 5B) and ESTIMATE (Figure 5C) algorithms to explore the immune infiltration in subgroups. A higher percentage of M2 macrophages was observed in the high-risk score group from the result of CIBERSORT, while T cells CD8+ accounted for a smaller percentage compared to the low-risk score group (Figure 5A, Supplementary Table 8). The ssGSEA algorithm showed a higher percentage of regulatory T cells (Tregs), as well as a lower percentage of activated CD8+ T cells and activated B cells in the high-risk group (Figure 5B, Supplementary Table 9). The ESTIMATE algorithm also confirmed the differences in immune infiltration between the high- and low-risk groups (Figure 5C). In the following work, we analyzed the connection between signature genes and immune cells. As shown in Figure 5D, VCAN had a significant correlation with M2 macrophages. GNG8 was positively correlated with Tregs and was negatively correlated with activated natural killer cells (Supplementary Table 10). Surprisingly, HLA gene expression and immune checkpoint gene expression varied significantly across the 2 risk groups (Figure 5E,F, Supplementary Tables 11,12).
To further characterize the effect of the ADCP-based signature on the TME, we examined the variations of immune pathways between the subgroups. The ssGSEA method was used to provide an estimated value for each cancer sample’s immune pathway, which is presented in Figure 6A and Supplementary Table 13. The association of 4 key genes in the signature with immune-related pathways was also explored (Figure 6B, Supplementary Table 14). A comparative study of significantly enriched pathways between subgroups was accomplished using GSEA, and the 5 most strongly associated pathways were selected for presentation (Figure 6C,D, Supplementary Table 15).
Drug sensitivity and immunotherapy efficacy prediction
As shown in Figure 7A, patients with high-risk scores exhibited a higher TIDE scoring. Nine compounds with sensitization differences in the high- and low-risk groups were illustrated. In the high-risk group of ADCP-based signatures, the IC50 of GNF.2, Z.LLNle.CHO, AP.24534, imatinib, NSC.87877, NVP.TAE684, pazopanib, X17.AAG, and PHA.665752 were significantly lower when compared with the low-risk group (Figure 7B).
Construction of nomogram
Univariate Cox analysis of the risk score and clinical parameters (age, gender, stage, and grade) demonstrated that age, stage, and risk scores could serve as prognostic factors for GC patients (Supplementary Fig. 2). With additional multivariate Cox regression analysis, a clinical risk model consisting of 3 independent prognostic factors: age, stage and risk score (Figure 8A) was finally constructed as a nomogram (Figure 8B). The Kaplan–Meier curve demonstrated that patients with high nomogram scores have a more unfavorable prognosis (Figure 8C; log-rank test, p = 1.77e-06). The AUC values for 1, 3 and 5 years shown in the ROC curves (Figure 8D) were 0.669, 0.675 and 0.685, respectively. Calibration curves (Figure 8E), as well as DCA curves (Figure 8F), were evaluated for the nomogram, implying it has a good level of predictive accuracy.
Discussion
Due to the heterogeneity of GC, the survival durations among patients exhibits huge variation, which covers a range from 5 months to 10 years.17, 18 Patients with early-stage localized GC have a 5-year OS rate above 60%, while in those diagnosed with distant metastasis, it is lower than 5%.19 Encouragingly, the exploration of reliable biomarkers through bioinformatics has demonstrated remarkable potential in clinical applications. It was recognized that prognostic signatures derived from multiple genes exhibited a significant role in the survival prediction of malignancies. We built an ADCP-based prognostic signature incorporating TCGA data and verified its practicability using the GEO dataset, as well as researching its characteristics with TME. The signature correlates with multiple immune cells and immune checkpoints. Additionally, differential drug sensitivities and immune efficacies were detected in the subgroups.
The 4 genes in the ADCP-based signature (MKNK2, VCAN, LRAT, and GNGB) have been reported in association with the progression of cancer. As in previous studies, MKNK2 (threonine kinase 2/MAP kinase interacting serine) has been considered an oncogene, which acts as a mediator of various cellular processes to promote the development of prostate cancer and gliomas.20, 21 Furthermore, it has been discovered that MKNK2 can be targeted by miR-125b, leading to the progression of breast cancer.22 A previous study also demonstrated that MKNK2 contributes to the enhancement of chemoresistance of ovarian cancer by inhibiting autophagy through miR-125b.23 Versican (VCAN) accumulates in tumor cells and mesenchyme as a protein and is regulated by cytokines. In cancer research, it has been proven that VCAN is involved in the progression of GC. Moreover, VCAN has been recognized as an independent prognostic predictor of GC.24 Lecithin retinol acyltransferase (LRAT) converts retinol to retinyl esters, regulating cell growth and differentiation.25 Researchers have demonstrated a significant loss of LRAT expression in invasive bladder cancer, correlating with an increasing tumor stage.26 G protein subunit gamma 8 (GNG8) participates in chemokine signaling that controls leukocyte migration across the endothelium, which may have a potential implication for the TME.27
As research on the TME deepens, the development of new immunotherapy regimens targeting the TME is becoming a major field of interest for cancer treatment. Given that the characteristics of the TME can influence the efficacy of immunotherapy, we focused on the role of ADCP signatures in the TME. The high-risk group of ADCP signatures exhibited a lower probability of survival, resulting from the suppression of the TME. As an important component of the TME, M2 macrophages can secrete undesirable cytokines, thus promoting tumor angiogenesis and tumor metastasis, which is detrimental to patient prognosis. A larger percentage of M2 macrophages was found in the high-risk group, while CD8 T cells, the main contributor that kills tumor cells, displayed a significant proportional decline. Interestingly, there are differential infiltrations of Tregs between the high- and low-risk groups. Tregs are key immune cells with immunosuppressive abilities in the TME, which exert effects through mechanisms such as secreting cytokines, limiting the activation of CD4+ helper T cells and CD8+ cytotoxic T cells, and regulating antigen-presenting cell (APC) functions.28, 29, 30, 31, 32, 33 This hinders GC patients from benefiting from ICI therapy. Targeting Tregs holds great potential for reshaping the GC immune microenvironment, enhancing anti-tumor immune responses and improving the OS rate of GC patients. This study stratified GC patients based on ADCP-related genes at the transcriptomic level and also stratified the abundance of Tregs. For GC patients with high Treg activity, traditional ICIs may not be sufficient to fully activate anti-tumor immune responses. In this case, adopting additional strategies to inhibit Treg activities becomes an effective supplementary approach.
Our functional enrichment analysis further suggested that the ADCP model regulates the TME through multiple immune pathways and has a strong immune characteristic. Considering high-risk patients may not benefit as much from immunotherapy, we screened for sensitive and effective compounds for high-risk patients and found that they are more sensitive to pazopanib and imatinib. Notably, imatinib and pazopanib are both tyrosine kinase inhibitors. In addition to the 2 targeted drugs, some chemical drugs were also identified; however, most are limited to cellular experiments. Moreover, traditional cancer chemotherapy based on chemical drugs is prone to drug resistance and toxic side effects. As an improved form of chemotherapy, metronomic chemotherapy involves administering low doses of drugs continuously without long breaks, which can help avoid these 2 drawbacks. More than 10 years ago, Chinese scholars demonstrated the efficacy and good tolerability of metronomic capecitabine for palliative treatment of advanced GC patients after fluoropyrimidine-based chemotherapy.34 Furthermore, the good tolerability and potential durable anti-tumor activity of metronomic capecitabine in patients with hepatocellular carcinoma undergoing sorafenib treatment was also confirmed.35 Mechanistically, metronomic chemotherapy works by inhibiting tumor angiogenesis and inducing immunogenic cell death (ICD), thereby regulating vascular–immune crosstalk.36 As a result, the combination of metronomic chemotherapy with ICIs has shown synergistic therapeutic effects in preclinical and clinical studies. Especially for high-risk patients, the combination of both may be an effective approach. Unfortunately, metronomic chemotherapy is currently used as a palliative standard care tool. Utilizing bioinformatic methods to identify biomarkers can benefit metronomic chemotherapy and may expand the role of metronomic chemotherapy in cancer therapy.
To more accurately exploit the predictive ability of the ADCP signature, we combined univariate and multivariate analysis with Cox regression and eventually constructed a nomogram selecting age, stage and risk scores. This nomogram visualized the logistic regression model to facilitate rapid clinical judgment on the prognosis of GC patients. The AUC curve demonstrated that the model predicted the prognosis of GC patients with reliable accuracy. The calibration curve illustrates that the predicted probability of patient survival is in good agreement with the actual probability, and its clinical application can be attempted.
Limitations
Despite the fact that this research made some contributions, it did have several shortcomings. First, although our work evaluated high sample sizes of GC cohorts to construct a well-validated prognostic signature, the use of diverse platforms might generate sampling bias, which may induce some ambiguity in the assessments of gene expression. Second, the underlying mechanisms by which the 4 genes (MKNK2, VCAN, LRAT, and GNGB) that were combined into the ADCP signature in our investigation contributed to GC progression, and the unfavorable outcome still remains unexplained; additional in-depth research into their biological functions might generate fresh targets and therapeutic strategies. Third, recognizing that these cohorts are merely retrospective, more prospective clinical studies are necessary to confirm our results. In particular, future research needs to evaluate the predictive function of the ADCP signature throughout both prognosis and the responsiveness to different kinds of treatment interventions. Finally, the accuracy of CIBERSORT and ssGSEA is limited by the representativeness of the training data and the assumptions of the algorithms themselves, leading to biases in assessing the levels of immune cell infiltration. These 2 algorithms mainly focus on the composition and proportion of immunocytes, with limited evaluation of their functional states. This calls for the adoption of more validated algorithms to analyze immunocytes and to compare algorithm performance through methods like cross-validation. Additionally, integrating multi-omics data, including gene expression, proteomics and metabolomics data, is essential for inferring the functional states of immune cells.
Conclusions
We identified 4 prognosis-associated genes that were related to OS and the TME in GC by also constructing a model with strong predictive effects. To the best of our knowledge, this report is the first effort to develop a prognostic signature of ADCP-related genes for GC. Our study offers a novel option for the diagnosis and prediction of GC and may contribute new biomarkers for the treatment of GC.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors. Therefore, no ethical approval or consent was required. No administrative permission and/or licenses were acquired for this study to access the original data used in this research.
Supplementary data
The Supplementary materials are available at https://doi.org/10.5281/zenodo.11065549. The package includes the following files:
Supplementary Fig. 1. Proportional hazards assumption based on Schoenfeld’s global and individual test. (A–D) The p-values of the Schoenfeld’s global test are all greater than 0.05 in gene selection using univariate Cox regression (A) and multivariate Cox regression (B), selection of ADCP-based signature and clinical features using univariate cox regression (C), and multivariate Cox regression (D).
Supplementary Fig. 2. Selection of ADCP-based signature and clinical parameters using univariate Cox regression.
Supplementary Table 1. Antibody-dependent cellular phagocytosis-related genes.
Supplementary Table 2. 531 differentially expressed antibody-dependent cellular phagocytosis-related genes.
Supplementary Table 3. Results of GO.
Supplementary Table 4. Results of KEGG.
Supplementary Table 5. Selection of antibody-dependent cellular phagocytosis-related genes with prognostic value using univariate Cox regression analysis.
Supplementary Table 6. Selection of antibody-dependent cellular phagocytosis-related genes with prognostic value using multivariate Cox regression analysis.
Supplementary Table 7. Node information of protein-protein interactions.
Supplementary Table 8. Wilcoxon test results of immunocyte infiltration between risk subgroups using CIBERSORT.
Supplementary Table 9. Wilcoxon test results of immunocyte infiltration between risk subgroups using ssGSEA.
Supplementary Table 10. Spearman correlation results of immunocyte infiltration and signature genes using CIBERSORT.
Supplementary Table 11. Wilcoxon test results of HLA gene expression between risk subgroups.
Supplementary Table 12. Wilcoxon test results of immune checkpoint expression between risk subgroups.
Supplementary Table 13. Wilcoxon test results of immune pathways between risk subgroups using ssGSEA.
Supplementary Table 14. Spearman correlation results of immune pathways and signature genes using ssGSEA.
Supplementary Table 15. GSEA results between risk subgroups.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)