















Abstract
Immunotherapy has revolutionized oncology; however, its efficacy remains limited by the immunosuppressive tumor microenvironment (TME). This editorial synthesizes recent advances demonstrating how rationally designed combination strategies – particularly those incorporating the transforming growth factor beta/programmed death-ligand 1 (TGF-β/PD-L1) bispecific antibody platform (YM101/BiTP) and the multi-cytokine-armed oncolytic virus VG161 – can overcome resistance mechanisms. By concurrently dismantling immunosuppressive networks, activating innate immunity and remodeling the TME, these approaches show superior preclinical activity across challenging tumor phenotypes. The integration of mechanistic insights with evolving biomarker-driven strategies heralds a new era of personalized combination immunotherapy.
Key words: immunotherapy, tumor microenvironment, antibodies, bispecific, oncolytic virotherapy
Introduction
The advent of immune checkpoint blockade (ICB) targeting the programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) axis has marked a paradigm shift in cancer therapy.1, 2 While a subset of patients experience durable responses, the overall response rate remains limited.3 This limitation is primarily due to the complex immunosuppressive mechanisms within the tumor microenvironment (TME).4, 5 The TME comprises a heterogeneous network of cellular and molecular components – including immunosuppressive factors such as tumor growth factor beta (TGF-β) and PD-L1, impaired antigen presentation, and physical or structural barriers that limit immune cell infiltration – that collectively hinder effective antitumor immune responses.
As a result, non-inflamed tumors – often classified as immune-excluded (T cells restricted to the stromal margin) or immune-desert (minimal T cell infiltration) – exhibit resistance to ICB monotherapy.6, 7 Overcoming this resistance requires combination strategies that not only relieve immunosuppression but also enhance immune activation.8 Recent preclinical studies have highlighted 2 promising modalities: bispecific antibodies targeting complementary immunosuppressive pathways, and oncolytic viruses engineered to deliver immunostimulatory payloads.9, 10, 11 This editorial focuses on how these approaches, particularly when combined, may reshape current strategies to overcome ICB resistance.
The immunosuppressive TME as the primary barrier
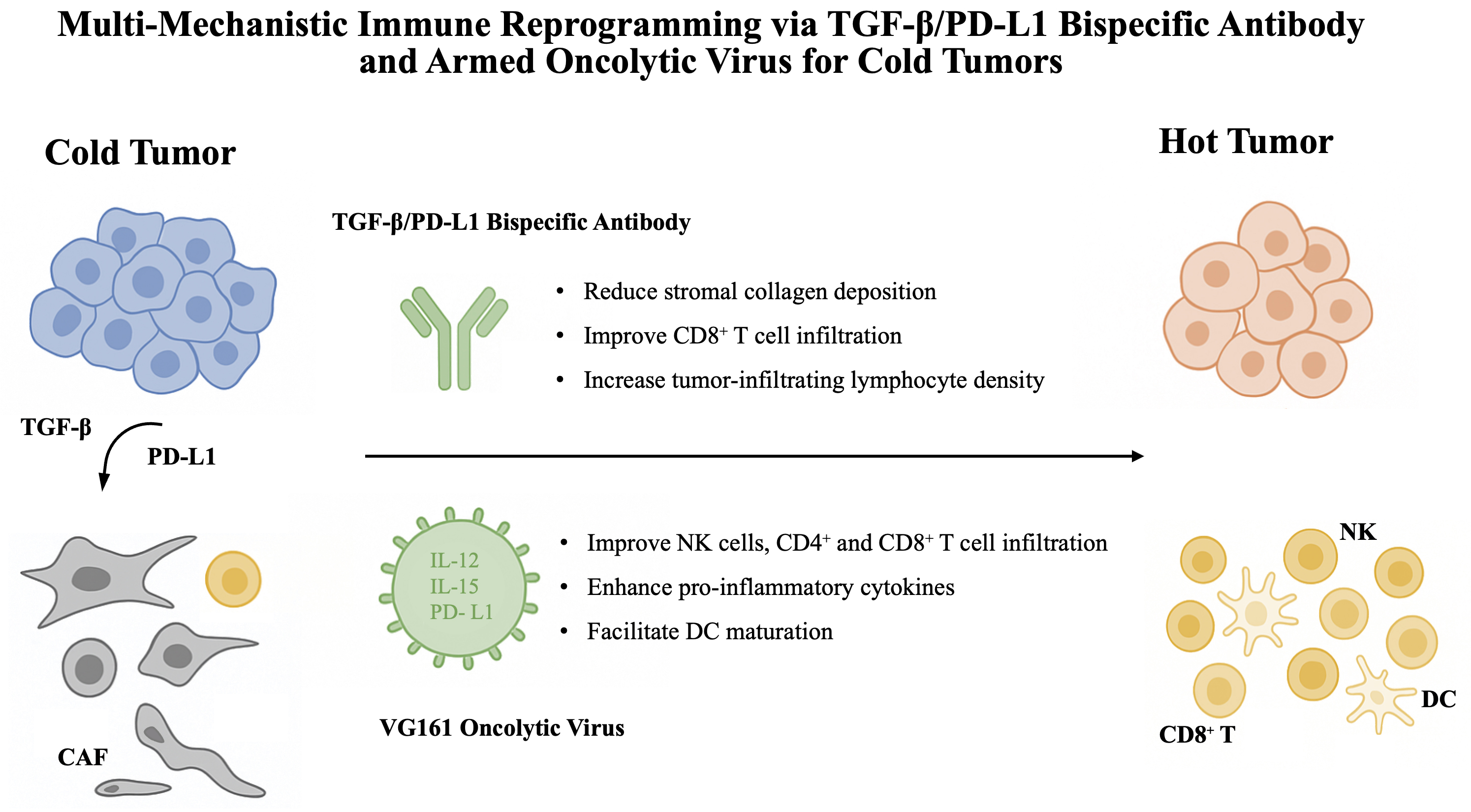
The TME functions as an immunosuppressive niche that actively inhibits antitumor immunity.12, 13, 14 Among the key mediators, TGF-β and PD-L1 signaling pathways play central roles. Transforming growth factor beta suppresses the effector function of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, promotes the differentiation of regulatory T cells (Tregs), induces cancer-associated fibroblast (CAF) activation, thereby generating desmoplastic stroma that limits T cell infiltration, and promotes the expansion of immunosuppressive myeloid populations.15, 16, 17, 18 Concurrently, PD-1/PD-L1 signaling impairs the function of activated T cells by inducing exhaustion, characterized by reduced cytokine production and proliferative capacity.19, 20 Therefore, the co-expression and spatial proximity of TGF-β and PD-L1 signaling contribute to a synergistic suppression of antitumor immunity.
In addition, defective innate immune activation – particularly the failure of immature dendritic cells (DCs) to present tumor antigens and prime T cells – underlies the immune-desert phenotype.6 Myeloid-derived suppressor cells (MDSCs) and M2-like tumor-associated macrophages (TAMs) further exacerbate immunosuppression by producing arginase, indoleamine 2,3-dioxygenase (IDO) and interleukin 10 (IL-10).21, 22, 23, 24, 25 This complex interplay between stromal, immune and tumor components results in T cell exclusion or dysfunction, thereby limiting the efficacy of ICB in the majority of patients.26 A comprehensive understanding of these mechanisms is critical for designing rational and effective combination therapies.
Combination therapy: A mechanistic approach to overcome resistance
Given the redundancy and compensatory nature of immunosuppressive pathways within the TME, monotherapies targeting individual molecules are insufficient to restore antitumor immunity. Combination approaches that simultaneously modulate multiple aspects of the cancer-immunity cycle offer a more effective strategy.8 Several therapeutic modalities have demonstrated potential in combination with ICB. Chemotherapy and radiotherapy promote immunogenic cell death, leading to antigen release and activation of DCs.27, 28, 29 Anti-angiogenic agents normalize tumor vasculature, reduce hypoxia and facilitate immune cell infiltration.30, 31 Co-stimulatory receptor agonists (e.g., CD40, OX40, GITR) enhance T cell activation and expansion.32 Agonists of the stimulator of interferon genes (STING) pathway activate type I interferon responses in DCs, promoting cross-priming of CD8+ T cells.33, 34, 35, 36 Epigenetic modulators reverse exhaustion-associated transcriptional programs in T cells,37 while metabolic regulators improve nutrient availability and reduce acidity in the TME, thus enhancing T cell viability and function.38, 39 In this context, bispecific antibodies that co-target non-redundant immunosuppressive pathways provide a spatially coordinated mechanism to inhibit immune evasion. These molecules offer dual blockade within the tumor site and may enhance efficacy while limiting systemic toxicity. 40 Ultimately, the effectiveness of combination regimens relies on mechanistic synergy – defined as the capacity to overcome multiple rate-limiting steps across antigen release, presentation, T cell priming, infiltration, and effector function.
VG161 oncolytic virus: Multi-mechanistic immune activation
Oncolytic viruses are a class of immunotherapeutic agents capable of inducing direct tumor cell lysis while stimulating innate and adaptive immunity.41 VG161 is a genetically modified oncolytic herpes simplex virus type 1 (oHSV-1) that encodes 3 immunostimulatory transgenes: IL-12, a single-chain IL-15/IL-15RA fusion protein and a PD-L1-targeting peptide.42 These payloads are designed to enhance local immune activation and counteract tumor-associated immunosuppression.
Upon intertumoral injection, VG161 selectively replicates in tumor cells, leading to immunogenic cell death and release of tumor-associated antigens (TAAs). This promotes the recruitment and activation of antigen-presenting cells, particularly DCs.11 The locally expressed IL-12 facilitates DC maturation and promotes Th1-type immune responses. The IL-15/IL-15RA fusion protein supports the survival and expansion of NK cells and CD8+ T cells, while the PD-L1-targeting fusion protein blocks inhibitory signaling in the TME.43, 44, 45
Preclinical studies in breast cancer models have demonstrated that VG161 increases the infiltration of CD4+ and CD8+ T cells, as well as NK cells, while enhancing the production of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interferon gamma (IFN-γ).11 Notably, VG161 exhibits strong synergy with paclitaxel (PTX). In addition to its cytotoxic effects, PTX facilitates antigen release and alters the TME to support viral replication and immune cell infiltration.46, 47 Sequential administration of VG161 followed by PTX results in enhanced tumor growth suppression, reduced pulmonary metastasis and increased CD3+ and CD8+ T cell infiltration in metastatic sites. These effects are more pronounced with VG161 than with its parental virus VG160, underscoring the importance of its immunomodulatory transgenes.
Bispecific targeting of TGF-β and PD-L1: From YM101 to BiTP
To overcome the limited efficacy of PD-1/PD-L1 blockade in immunosuppressive microenvironment, especially in immune-excluded tumors, a bispecific antibody strategy targeting both TGF-β and PD-L1 has been developed. YM101, constructed using the Check-BODY™ platform, combines binding domains for TGF-β and PD-L1 in a single molecule.10 Preclinical studies showed that YM101 effectively inhibited TGF-β–Smad and PD-L1–PD-1 signaling pathways, reversed epithelial–mesenchymal transition (EMT) and enhanced T cell activation in vitro. In murine tumor models, YM101 exhibited superior antitumor activity compared to monotherapy. This was accompanied by increased infiltration of CD8+ T cells and dendritic cells, a higher M1/M2 macrophage ratio, and enhanced cytokine production, collectively promoting a ‘hot’ tumor phenotype.
Building on these results, a humanized version of YM101 – termed BiTP – was developed to enable translational application.9 BiTP retained high binding affinity and functional activity against both TGF-β and human PD-L1. In humanized TNBC models, BiTP exhibited enhanced antitumor efficacy over anti-PD-L1 or anti-TGF-β monotherapy. Mechanistically, BiTP reduced stromal collagen deposition, improved CD8+ T cell infiltration and increased tumor-infiltrating lymphocyte density. These changes contributed to immune reprogramming within the TME and reinforced antitumor immunity. Together, YM101 and BiTP exemplify a promising bispecific antibody approach that simultaneously alleviates immune exclusion and checkpoint-mediated suppression, offering a novel therapeutic strategy for immune-excluded tumors.
Future perspectives
Current limitations of immune checkpoint blockade highlight the essential role of the immunosuppressive TME in mediating therapeutic resistance. Accumulating preclinical evidence supports the need for combination strategies that concurrently target multiple immunosuppressive mechanisms within the TME. In our recent studies, we employed the TGF-β/PD-L1 bispecific antibody platform and the cytokine-armed oncolytic virus VG161 as complementary strategies to address TME-associated immune exclusion and desert-like features.
The therapeutic efficacy of these platforms is further enhanced when used in combination with agents targeting innate immune activation or adaptive resistance. For example, YM101 demonstrates improved antitumor activity when combined with STING agonists, which enhance antigen presentation and type I interferon responses within the TME.36, 48 In addition, resistance-associated features such as CCR5+ T cell enrichment – identified through single-cell RNA sequencing – may be targeted by agents such as Maraviroc to further optimize therapeutic response.49 VG161 also exhibits synergy with chemotherapeutic agents such as paclitaxel, which not only induces direct cytotoxicity but also modulates suppressive myeloid cells, thereby amplifying systemic antitumor immunity and inhibiting metastatic dissemination.
To facilitate successful clinical translation, several key factors must be addressed. First, the development and validation of predictive biomarkers are critical for stratifying patients according to TME immunophenotypes (inflamed, excluded, desert) and dominant resistance mechanisms. This will enable rational selection of personalized combination regimens. Second, optimization of treatment sequencing and dosing is required to balance efficacy with toxicity. Third, ongoing exploration of novel combination strategies – including integration of bispecific antibodies or oncolytic viruses with metabolic modulators, epigenetic therapies, co-stimulatory receptor agonists, or adoptive cell therapies – holds considerable therapeutic potential.50 Finally, advances in spatial and single-cell multi-omics technologies are expected to provide high-resolution insights into TME dynamics during treatment, facilitating the identification of emergent resistance pathways and novel therapeutic targets.51
Conclusions
The integration of multi-targeted agents such as YM101/BiTP and VG161, informed by mechanistic insights and supported by biomarker-driven patient selection, represents a rational and promising approach to overcoming the immunosuppressive TME. These strategies may substantially broaden the clinical benefit of immunotherapy across a wider range of tumor types and patient populations.
Use of AI and AI-assisted technologies
Not applicable.