
















Abstract
Background. Liver cancer is a malignant tumor commonly seen in infants and young children. Serabelisib is a novel and effective phosphoinositide-3-kinase (PI3Kα) inhibitor, currently in trials for solid malignancy treatment, such as bladder cancer. However, it is unclear whether serabelisib affects liver cancer.
Objectives. To explore the effects of serabelisib on the proliferation, apoptosis, invasion, and metastasis of HepG2 and HuH-6 cells, and elucidate the relevant molecular mechanisms.
Materials and methods. The HepG2 cells were treated with 2 μM, 4 μM or 8 μM serabelisib, while the 0-μM group was used as a control. A plate clone formation assay was utilized to measure colony formation ability in each group, a 5-ethynyl-2’-deoxyuridine (EdU) assay examined cell proliferation, a Cell Counting Kit-8 (CCK-8) assay assessed cell viability, flow cytometry measured the cell cycle and apoptosis, JC-1 staining determined mitochondrial membrane potential, and transmission electron microscopy evaluated cell morphology. In addition, gene and protein expression levels of apoptosis markers, epithelial–mesenchymal transition (EMT), Gasdermin D (GSDMD), and the PI3K/protein kinase B (AKT) signaling pathway were measured.
Results. After serabelisib intervention, HepG2 and HuH-6 cells formed fewer colonies, proliferated more slowly, and had reduced viability. The number of HepG2 and HuH-6 cells in the G2 and S phases decreased, apoptosis and the number of apoptotic bodies increased, and mitochondrial membrane potential decreased. Serabelisib treatment also reduced the migration and invasion capacity of the cells. Furthermore, genes and proteins of the PI3K/AKT signaling pathway were downregulated, while those that promote apoptosis and pyroptosis or inhibit EMT were upregulated.
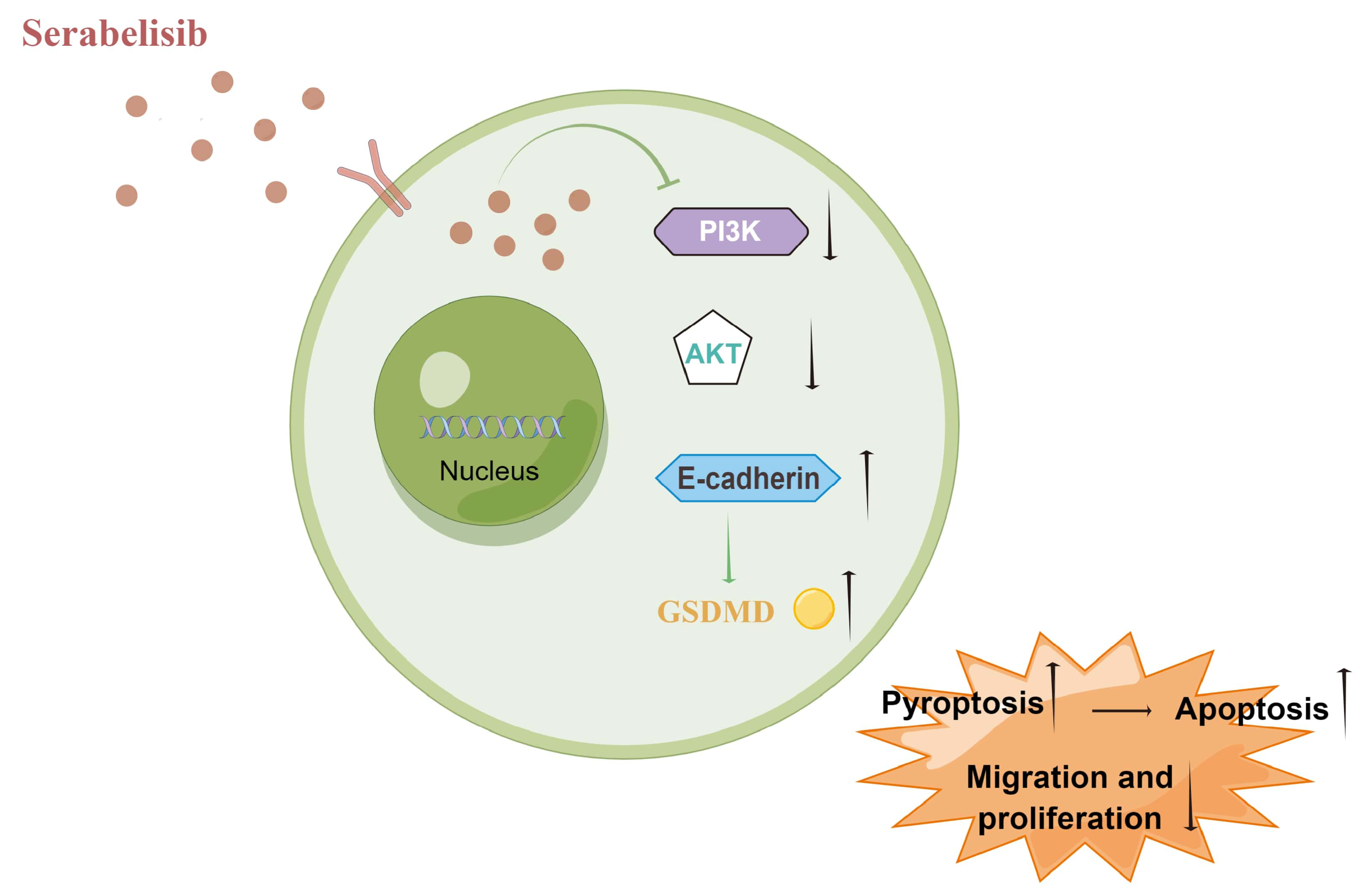
Conclusions. Serabelisib inhibited the PI3K/AKT signaling pathway, thereby inhibiting EMT and promoting apoptosis and pyroptosis in HepG2 and HuH-6 cells.
Key words: PI3K, hepatoblastoma, EMT, pyroptosis, serabelisib
Background
Liver cancer is a common malignant tumor that poses a grave threat to human life.1 Eighty percent of liver cancers in children and adolescents are primary hepatoblastomas.2 Chemotherapy and surgery are the primary treatments for hepatoblastoma,3 with cure dependent on complete excision. However, according to surgical guidelines, 60–80% of patients have nonresectable tumors at diagnosis.4 Therefore, researchers seek a pharmacological solution to liver cancer treatment.
Serabelisib (INK-1117, MLN-1117, TAK-117) is a novel, highly selective and effective phosphoinositide-3-kinase (PI3Kα) inhibitor administered orally.5 The drug is currently being explored for solid malignancy treatment and has shown promising results.6 In addition, combining serabelisib with TAK-228 has demonstrated synergistic anti-tumor effects in preclinical bladder cancer models.7 Meanwhile, serabelisib pharmacokinetics continue to improve.8 However, the effects of serabelisib on liver cancer have not been reported.
The PI3K/protein kinase B (AKT) pathway is a classical signal transduction pathway that promotes proliferation and suppresses apoptosis. It also participates in the growth promotion and proliferation of cancer cells, promotes cellular invasion and metastasis, enhances tumor angiogenesis, and induces apoptosis resistance to chemotherapy and radiotherapy.9 Hartmann et al. found that the PI3K/AKT signaling pathway is usually activated in liver cancer, and in vitro PI3K targeting leads to increased apoptosis, which inhibits liver cancer cell proliferation.10 Furthermore, many substances, such as JTC-801, have been shown to inhibit HepG2 metastasis through the regulation of PI3K/AKT signaling pathway.11
The PI3K/AKT signaling pathway can regulate epithelial–mesenchymal transition (EMT), a process in which epithelial cells lose their characteristics and acquire a mesenchymal phenotype under certain conditions, to reduce tumor aggression.12, 13 It has been reported that deoxyribonucleic acid (DNA) damage-regulated autophagy modulator 1 (DRAM1) regulates liver cancer cell migration and invasion through the autophagy–EMT pathway,14 while other studies demonstrated the PI3K/AKT involvement in renal cancer cell pyroptosis.15 Pyroptosis is a form of cell death that leads to inflammation and further damage to the body.16 Several studies have found many factors related to pyroptosis, including NLRP3, interleukin (IL)-18, IL-1β, apoptosis-associated speck-like protein containing a CARD (ASC), and caspase-1. In addition, interactions between caspase-1 and Gasdermin D (GSDMD) can lead to pyroptosis.17 Therefore, we hypothesized that serabelisib could interfere with HepG2 and HuH-6 pyroptosis through GSDMD regulation of the PI3K/Akt/E-cadherin signaling pathway.
Objectives
The study aimed to assess the effects of serabelisib on the PI3K/AKT signaling pathway in HepG2 and HuH-6 cells by exploring its impact on proliferation, apoptosis, invasion, and metastasis, and to elucidate the relevant molecular mechanisms. This research may provide a novel avenue for liver cancer treatment by establishing a new experimental and theoretical basis for the clinical study of serabelisib in liver cancer.
Materials and methods
Drug treatment
The HepG2, HuH-6, SMMC-7721, and human kidney fibroblast (HKF) cells were purchased from Abiowell (Changsha, China). The cells were digested with 0.25% trypsin solution (C0201; Beyotime Biotechnology, Shanghai, China), and cell suspensions were prepared using Roswell Park Memorial Institute (RPMI) 1640 complete medium (04-001-1ACS; Thermo Fisher Scientific, Waltham, USA). Cells were inoculated on a 6-well plate at approx. 2×105 cells per well and cultured overnight in a DH-160I incubator (Shanghai Sanotac Scientific Instruments Co., Ltd., Shanghai, China) at 37°C with saturated humidity and 5% carbon dioxide. The small-molecule inhibitor serabelisib (S8581; Selleckchem, Houston, USA) was dissolved in sterile dimethyl sulfoxide (DMSO) and diluted to the desired concentration. The cells in each group were treated with different drug interventions. Cells in the 0-μM group were cultured without intervention, cells in the 2-μM group were cultured with a medium containing 2 μM of serabelisib, 4 μM of serabelisib was added to the medium used to culture cells in the 4-μM group, and the 8-μM group had 8 μM of serabelisib added to the medium. For the serabelisib+MK2206 group, 8 μM of serabelisib and 3 μM of MK2206 (IM1090; Solarbio, Beijing, China) were added to the medium. The intervention time was 48 h.
Plate clone formation assay
After 24 h of intervention, the cells were digested with a 0.25% trypsin solution and added to high-sugar Dulbecco’s modified Eagle’s medium (DMEM) (D5796-500mL; Thermo Fisher Scientific) to prepare cell suspensions. Then, the cells were inoculated onto a 6-well plate at a density of 1000 cells per well and shaken thoroughly.18 Afterwards, they were cultured in an incubator (37°C, 5% CO2) for 7 days. The colony-forming ability was observed under an inverted biological microscope (model DSZ2000X; Cnmicro, Beijing, China).
5-ethynyl-2’-deoxyuridine assay
After 24 h of intervention, the 5-ethynyl-2’-deoxyuridine (EdU) DNA Proliferation in vitro Detection Kit (C10310; RiboBio, Guangzhou, China) assessed the HepG2 proliferation rate. In brief, the cells were incubated in a medium containing 50 μM of EdU for 24 h, incubated with 1× Apollo® staining solution and then stained with 1× Hoechst33342 solution. Observations were made immediately after staining.
Cell Counting Kit-8 assay
The medium was aspirated, discarded and replaced with 110 μL of Cell Counting Kit-8 (CCK-8) (NU679; Dojindo Molecular Technologies, Rockville, USA) working solution which consisted of 100 μL of high-sugar DMEM and 10 μL of CCK-8 stock solution. Cells were then cultured for another 4 h in a 37°C and 5% CO2 incubator. Finally, the absorbance of each well was measured at 450 nm using a microplate analyzer (model MB-530; Shenzhen Heales Technology Development Co., Ltd., Shenzen, China).
Flow cytometry (cell cycle)
The cells were gently suspended and separated into single cells. Then, 1.2 mL of pre-cooled 100% ethanol was added drop by drop until the concentration reached 75%. The mixed solution was stored at 4°C overnight for fixing. The next day, the cells were washed 3 times and incubated with 150 μL of propidium iodide (PI) (MB2920; Dalian Meilunbio, Dalian, China) in the dark at 4°C for 30 min. After incubation, the cells were transferred to the flow testing tube and measured using CytoFLEX A00-1-1102 instrument (Beckman Coulter, Brea, USA).
Flow cytometry (cellular apoptosis)
An apoptosis kit (KGA108; KeyGEN Bio, Nanjing, China) was used to assess cell apoptosis. Each 2×105 aliquot was suspended in 500 μL of binding buffer, and 5 μL of annexin V-fluorescein isothiocyanate (FITC) was added to the cell suspension and mixed well. The mixture was then incubated with 5 μL of PI in the dark at room temperature for 10 min.
JC-1 staining
A mitochondrial membrane potential assay kit with JC-1 (C2006; Beyotime Biotechnology) detected early cell apoptosis. Each 50-μL JC-1 (200×) aliquot was diluted with 8 mL of ultra-pure water and mixed to obtain the JC-1 staining working solution. After incubation, the cells were washed twice with JC-1 staining buffer (1×) and photographed for observation.
Scratch assay
A flask of cells was digested with trypsin, counted and used to inoculate 6-well plates at 5×105 cells/well. When cells reached 90% confluency, they were scratched with a pipet tip, unattached cells were washed with phosphate-buffered saline (PBS), and serum-free high-sugar DMEM (D5796; Thermo Fisher Scientific) was added. The scratches were photographed and recorded at 0 h and 24 h.
Transwell migration assay
Cell suspensions were prepared using serum-free basal medium, added to the upper chambers of transwell plates (33318035; Corning Inc., Corning, USA) and incubated for 48 h. Then, the cells were immobilized for 20 min in an acetone and methanol solution (Sinopharm, Beijing, China) mixed at a 1:1 ratio. The cells were stained with 0.5% crystal violet (C8470; Solarbio) for 5 min and washed with water. Cells on the external surface were observed and imaged using an inverted microscope (model DSZ2000X; Cnmicro).
Transwell invasion assay
Sixty mililiters of Matrigel solution (356231; Corning) were diluted in 120 μL of serum-free medium, and 60 μL of diluted matrix glue was added to each transwell. The transwells were placed in an incubator (37°C, 60 min) to solidify the gel. Residual liquid was aspirated, 70 μL of a basic medium was added to each transwell, and the basement membrane was hydrated at 37°C for 30 min. The cell suspension was prepared with a basic serum-free medium containing bovine serum albumin (BSA) (5 g/L), and cell density was adjusted to 1×105/mL. After 48 h, the Matrigel glue and cells on the upper compartment were wiped. Finally, the membranes were observed and imaged using an inverted microscope (model DSZ2000X; Cnmicro).
Immunofluorescence
Total ribonucleic acid (RNA) was extracted using TRIzol lysate (15596026; Thermo Fisher Scientific). The protocol included 0.3% Triton permeabilization for 30 min and 5% BSA blocking for 60 min at 37°C. Appropriately diluted primary antibodies, including GSDMD (20770-1-AP, 1:50; Proteintech, Rosemont, USA) and caspase-1 (22915-1-AP, 1:50; Proteintech), were added dropwise at 4°C overnight. Cells were then fixed in 4% paraformaldehyde for 10 min. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) working solution (AWI0331a; Abiowell) at 37°C for 10 min.
Western blot
Total cell protein was extracted using radioimmunoprecipitation (RIPA) lysate (P0013B; Beyotime Biotechnology). Separation gels with concentrations of 10%, 12% and 4.8% were prepared. After closure, membranes were incubated with primary PI3K (4249, 1:1000; Cell Signalling Technology, Danvers, USA), N-cadherin (22018-1-AP, 1:2000; Proteintech), p-AKT (4060, 1:2000; Cell Signalling Technology), E-cadherin (20874-1-AP, 1:5000; Proteintech), cytochrome C (ab184699, 1:5000; Abcam, Cambridge, UK), caspase-9 (#20750, 1:1000; Cell Signalling Technology), caspase-3 (9664, 1:1000; Cell Signalling Technology), B-cell lymphoma 2 (Bcl-2) (12789-1-AP, 1:2000; Proteintech), GSDMD (66387-1-Ig, 1:10000; Proteintech), NLRP3 (19771-1-AP, 1:800; Proteintech), caspase-1 (ab179515, 1:1000; Abcam), ASC (ab180799, 1:1000; Abcam), IL-1β (16806-1-AP, 1:2000; Proteintech), IL-18 (ab191860, 0.25 µg/mL; Abcam), and β-actin (66009-1-Ig, 1:5000; Proteintech) antibodies for 90 min. After incubation, the membrane was washed 3 times with PBS Triton X (PBST) solution.
Statistical analyses
All data are expressed as mean ± standard deviation (M ±SD). The analysis employed IBM Statistical Package for Social Sciences (SPSS) v. 26.0 software (IBM Corp., Armonk, USA). The Kolmogorov–Smirnov test analyzed data distribution, and all data displayed a normal distribution and homogeneity of variance. The data were analyzed using parametric tests, with one-way analysis of variance (ANOVA) and Tukey’s post hoc test used to compare data among the 3 groups. Statistical significance was set at p < 0.05.
Results
Screening cell lines
First, we examined the effects of serabelisib on HepG2, HuH-6, SMMC-7721, and HKF cell proliferation. The results of the CCK-8 experiments showed that the proliferation ability of HepG2 cells in the 2-μM, 4-μM and 8-μM groups significantly decreased compared to the 0-μM group (Figure 1A). Compared with the 0-μM group, the proliferation ability of HuH-6 cells was significantly attenuated at 4 μM and 8 μM, but not at 2 μM (Figure 1B). The proliferation of SMMC-7721 cells was moderated at 2 μM, 4 μM and 8 μM, but not to a statistically significant extent (Figure 1C). The proliferation of HKF cells was not altered by serabelisib treatment (Figure 1D). The raw data and analysis are presented in Supplementary Table 1. Serabelisib had no effect on normal HKF cells. Based on these findings, we selected HepG2 and HuH-6 for subsequent experiments.
Serabelisib affected the PI3K/Akt/
E-cadherin signaling pathway and pyroptosis
To further understand the effects of serabelisib on cells at the gene and protein levels, western blot was used to detect apoptosis and EMT-related factors in HepG2 and HuH-6 cells. Compared with the 0-μM group, the intracellular expression levels of PI3K and p-AKT were significantly decreased in the 4-μM and 8-μM groups. After serabelisib intervention, the expression of Bcl-2 was decreased, while the expressions of cytochrome C, caspase-9 and caspase-3 were upregulated. Moreover, N-cadherin expression decreased, and E-cadherin expression increased in the drug intervention group, suggesting that serabelisib can affect the expression of apoptosis and EMT-related factors (Figure 2A). We used immunofluorescence to analyze the fluorescence intensity of caspase-1 and GSDMD, and explore whether serabelisib affects pyroptosis-related factors. The results indicated that caspase-1 and GSDMD fluorescence intensity increased at 4 μM and 8 μM compared to the 0-μM group (Figure 2B). Western blot was used to detect the expression of pyroptosis-related proteins. The results showed increased pyroptosis-related expression of GSDMD, GSDMD-N, NLRP3, caspase-1, ASC, IL-1β, and IL-18 (Figure 2C). All data are presented in Supplementary Table 2.
In short, serabelisib affected the PI3K/Akt/E-cadherin signaling pathway and pyroptosis-related protein expression.
Serabelisib affected the proliferation and apoptosis of HepG2 and HuH-6 cells through the PI3K/Akt/E-cadherin signaling pathway
We examined the effects of serabelisib on the proliferation and apoptosis of HepG2 and HuH-6 cells. Plate clone formation and EdU experiments showed that serabelisib reduced the number of cloned colonies and weakened proliferation ability. After adding the AKT signaling pathway inhibitor MK2206, the colony-forming ability of cells was significantly reduced (Figure 3A,B). To investigate whether serabelisib can promote apoptosis, flow cytometry examined apoptosis and cell cycle distribution. As shown in Figure 3C,D, the proportion of apoptotic cells increased, and the proportion of cells in the G2 and S phases decreased after the treatment with serabelisib and MK2206 in HepG2 and HuH-6 cells. Furthermore, serabelisib and MK2206 gradually increased the number of apoptotic cells, and JC-1 changed from a red polymer to a green monomer (Figure 3E). The data and results are presented in Supplementary Table 3. Briefly, serabelisib and MK2206 can promote apoptosis and inhibit the proliferation of HepG2 and HuH-6 cells.
Serabelisib affected the migration of HepG2 and HuH-6 cells through the PI3K/Akt/E-cadherin signaling pathway
Our findings indicated that serabelisib could affect the proliferation and apoptosis of liver cancer cells through the PI3K/Akt/E-cadherin signaling pathway. The scratch method was employed to explore whether serabelisib could alter HepG2 and HuH-6 migration. Compared with the control group, the horizontal migration ability of the cells in the serabelisib group was reduced, while the horizontal migration ability was significantly reduced in the serabelisib+MK2206 group compared to the serabelisib group (Figure 4A).
Transwell assays assessed the ability of cells to invade and migrate vertically. As shown in Figure 4B,C, the migratory and invasive abilities of the cells in the serabelisib group were significantly reduced compared with the control group. Moreover, serabelisib+MK2206 treated cells displayed less migration and invasion capacity than the cells in the serabelisib group. The raw data and results for Figure 4 are presented in Supplementary Table 4. In summary, serabelisib inhibited migration and invasion of HepG2 and HuH-6 cells.
Serabelisib affected GSDMD-mediated pyroptosis in HepG2 and HuH-6 cells through the PI3K/Akt/E-cadherin signaling pathway
The results indicated that serabelisib affected liver cancer cell migration and invasion. We explored whether serabelisib could affect the pyroptosis of HepG2 and HuH-6 cells using immunofluorescence to detect the pyroptosis-related caspase-1 and GSDMD. Compared with the control group, the fluorescence intensity of caspase-1 and GSDMD increased in the serabelisib group, while the fluorescence intensity increased sharply in the serabelisib+MK2206 group compared to the serabelisib group. These findings indicate that PI3K/Akt/E-cadherin signaling axis inhibition by serabelisib could increase pyroptosis-related gene expression levels (Figure 5A).
Western blot analysis of the pyroptosis-related pathway showed increased GSDMD, NLRP3, caspase-1, ASC, IL-1β, and IL-18 protein levels in the serabelisib group compared to the control group. Furthermore, GSDMD, NLRP3, caspase-1, ASC, IL-1β, and IL-18 significantly increased in the serabelisib+MK2206 group compared with the serabelisib group (Figure 5B). All raw data and analysis are presented in Supplementary Table 5. The results indicate that PI3K/Akt/E-cadherin inhibition using serabelisib can promote pyroptosis-related gene expression in cells and trigger pyroptosis.
Discussion
Liver cancer originates from abnormal stem cells and hepatic epithelial progenitor cells,19 and is a malignant embryonic liver tumor commonly found in infants and young children, accounting for around 80% of pediatric liver cancer cases.19 Currently, liver cancer therapy includes adjuvant chemotherapy, hepatectomy and liver transplantation. Unfortunately, the survival rate is only 60% for high-risk patients.20 Serabelisib is an effective oral selective PI3Kα inhibitor currently being explored for clinical use in cancer treatment.21 In this study, serabelisib was tested in vitro to assess a range of cell functions. We found that serabelisib promoted apoptosis in a dose-dependent manner. To further explain its mechanism of action, we examined the cellular gene and protein levels.
The PI3K/AKT axis contributes to oncogenic transformation, and its possible mechanisms include stimulation of proliferation, metastasis, and the inhibition of autophagy and senescence.22 Apoptosis is a form of programmed cell death regulated by proteins of the Bcl-2 and caspase families,23 with Bcl-2 being an anti-apoptotic gene and an important target for cancer therapy.24 The best biochemical marker recognized for early and late apoptosis detection is cysteine protease activation.25 During apoptosis, caspase-3 acts as an executor and has an important proteolytic function, regulating the final stage of programmed cell death.26 According to reports, lncRNA prostate cancer-associated transcript 1 (PCAT1) interacts with dyskerin pseudouridine synthase 1 to regulate proliferation, invasion and apoptosis in non-small cell lung cancer cells via the vascular endothelial growth factor (VEGF)/AKT/Bcl-2/caspase-9 pathway.27 We found that serabelisib inhibited expression of PI3K, AKT and Bcl-2 in cells, while the expression of cytochrome C, caspase-9 and caspase-3 was elevated. In summary, serabelisib promotes HepG2 and HuH-6 apoptosis by inhibiting the PI3K/AKT signaling pathway.
Tumor metastasis and recurrence are key barriers to a complete liver cancer cure.28 Epithelial–mesenchymal transition is necessary for promoting migration and invasion of tumor cells, and is often characterized by decreased epithelial E-cadherin and increased N-cadherin.29, 30 The EMT of hepatocellular carcinoma (HCC) is considered crucial in intrahepatic dissemination and distal metastasis.31 During EMT, epithelial cells lose their polygonal shape and acquire a fusiform conformation, resulting in enhanced tumor cell motility and invasion abilities.32 In HCC, the protein arginine methyltransferase activates EMT by regulating the PI3K/AKT signaling pathway to promote HCC cell invasion and lung metastasis.33 In this study, the results indicate that PI3K, p-AKT and E-cadherin expression levels in HepG2 and HuH-6 cells decreased after serabelisib intervention, while the expression of N-cadherin increased, and the effect was dose-dependent.
Limitations
Our future research will focus on investigating the effects of serabelisib on tissue growth in xenotransplantation tumor models. The current study demonstrated that serabelisib inhibited liver cancer development in a dose-dependent manner and explained its molecular mechanisms, providing a theoretical basis for its formal application in the clinical treatment of liver cancer. However, this study only involved mouse models and did not include a wider population or clinical trials. Therefore, further research is needed to determine the potential and efficacy of serabelisib in treating liver cancer.
Conclusions
Serabelisib inhibited cell migration and invasion by blocking EMT through the PI3K/AKT signaling pathway. Remarkably, GSDMD, a major pyroptosis effector, was repressed by serabelisib. We believe that pyroptosis plays a vital role in liver cancer and is inhibited by serabelisib. Serabelisib inhibited apoptosis and pyroptosis at the gene and protein levels.
Supplementary data
The supplementary materials are available at https://doi.org/10.5281/zenodo.7964600. The package contains the following files:
Supplementary Table 1. Proliferation ability of HepG2, HuH-6, SMMC-7721, and HKF cells in the 2-μM, 4-μM and 8-μM groups (for Figure 1).
Supplementary Table 2. Expression of PI3K/Akt/E-cadherin signaling pathway and pyroptosis (for Figure 2).
Supplementary Table 3. Proliferation and apoptosis of HepG2 and HuH-6 cells (for Figure 3).
Supplementary Table 4. Migration of HepG2 and HuH-6 cells through the PI3K/Akt/E-cadherin signaling pathway (for Figure 4).
Supplementary Table 5. Pyroptosis of GSDMD-mediated HepG2 and HuH-6 cells through the PI3K/Akt/E-cadherin signaling pathway (for Figure 5).




