
















Abstract
Background. Ferroptosis is a type of iron-dependent programmed cell death. The inhibition of ferroptosis has been reported to alleviate myocardial ischemia/reperfusion injury (IRI). However, it is unknown whether this protective effect occurs in the ischemia or reperfusion phase. Sestrin 1 (Sesn1) possesses remarkable cytoprotective functions to diverse cellular stresses. However, whether Sesn1 is involved in the regulatory process of ferroptosis during myocardial IRI is unknown.
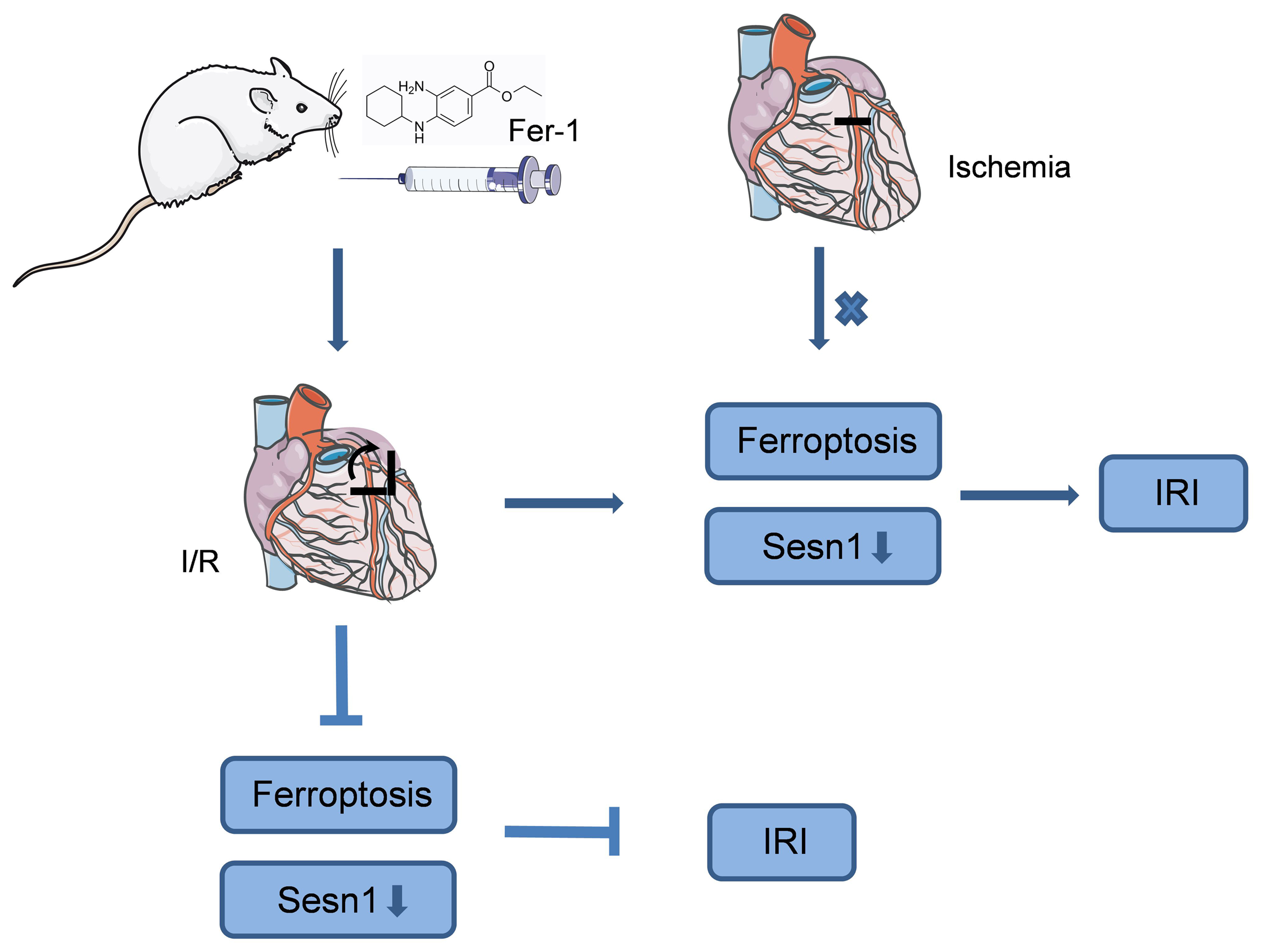
Objectives. This study aimed to simulate an acute myocardial infarction (AMI) that occurs in rats within 6 h, verify the occurrence and effects of ferroptosis in the phases of ischemia and reperfusion, and further explore the relationship between ferroptosis, IRI and Sesn1.
Materials and methods. The hearts of Sprague Dawley (SD) rats undergoing ischemia for varying lengths of time or having undergone ischemia followed by varying lengths of reperfusion were examined. The occurrence of ferroptosis was verified by detecting changes in ferroptosis biomarkers. In addition, ferrstatin-1 (Fer-1) was administered to demonstrate the effect of ferroptosis in myocardial IRI and to detect changes in Sesn1.
Results. The results showed that the myocardial damage was more severe with more prolonged myocardial ischemia. There were no significant changes in ferroptosis biomarkers in cardiac tissues during the ischemia phase, the levels of iron and malondialdehyde (MDA) were elevated, and the expression of glutathione peroxidase 4 (GPX4) and ferritin heavy chain 1 (FTH1) were decreased after myocardial IRI. Compared to the ischemia/reperfusion (I/R) group, the treatment with Fer-1 before reperfusion can attenuate myocardial IRI, reverse the decrease in GPX4 and FTH1 expression, and decrease the rise in iron content and MDA. In addition, we found that the expression of Sesn1 was reduced in hearts that suffered IRI; however, the treatment with Fer-1 can reverse this situation.
Conclusions. Ferroptosis occurred during the myocardial reperfusion phase but not ischemia. The inhibition of ferroptosis exerted beneficial effects on myocardial IRI, providing a theoretical basis for targeted therapy in patients with AMI. Sestrin 1, regulated by ferroptosis, may play an important role in myocardial IRI.
Key words: ferroptosis, myocardial IRI, Fer-1, Sesn1
Background
Acute myocardial infarction (AMI) has become one of the most dangerous diseases worldwide. The high medical costs and complications severely affect the quality of life. Currently, reperfusion strategies, such as percutaneous coronary intervention (PCI), are recommended as the optimal treatment for AMI.1 The treatment within the first 6 h is considered optimal. With the development of interventional therapy and the rise in people’s awareness, the treatment rate of patients with AMI within 6 h has improved, meaning that patients are receiving timely treatment. However, the consequent reperfusion will likely precipitate paradoxical cardiomyocyte dysfunction, namely ischemia/reperfusion injuries (IRIs). Studies in animal models of AMI suggested that lethal reperfusion injury accounted for up to 50% of the final size of the myocardial infarct.2 Several preventive strategies have been proposed in these models to ameliorate reperfusion injuries. However, the results were disappointing. Over the past decade, researchers have proposed that many mechanisms contribute to IRI, such as calcium overload, oxidative stress, inflammation, and energy metabolism disorders, ultimately leading to myocardial cell death.3
Previously, apoptosis and necrosis were considered the main types of cardiomyocyte death. Apoptosis is a form of programmed cell death, while necrosis is a form of an accidental and uncontrolled pathological cell death. Recently, studies have demonstrated that necrosis can be regulated by different signaling pathways, namely regulated cell death (RCD).4, 5 According to the recommendations of the 2018 Nomenclature Committee on Cell Death (NCCD), RCD can take various forms, such as necrotic apoptosis, pyroptosis and ferroptosis, among others.6 Ferroptosis is a relatively recently defined form of iron-dependent programmed cell death, first proposed by Dixon et al. in 2012.7 According to the existing literature, ferroptosis is characterized by 2 aspects that distinguish it from apoptosis, necroptosis and autophagy. Regarding cell morphology, ferroptosis reduces cell mitochondria, increases mitochondrial membrane density and decreases mitochondrial cristae. Regarding cell composition, ferroptosis results in lipid peroxidation and the accumulation of reactive oxygen species (ROS). Iron is an essential mineral involved in different biological processes within organisms. Iron metabolism disorders participate in the pathological processes associated with many diseases, including type 2 diabetes, obesity, nonalcoholic fatty liver disease, and cardiovascular disease.8, 9, 10 Ferroptosis leads to lipid peroxidation and high levels of ROS. Oxidative stress was one of the mechanisms involved in myocardial injury. Therefore, we wanted to elucidate whether ferroptosis was involved in myocardial ischemia and/or reperfusion injuries.
Sestrin 1 (Sesn1) belongs to the sestrin family, a group of highly conserved, stress-induced proteins that participate in various pathophysiological processes.11, 12, 13, 14, 15 The p53 tumor suppressor protein induces the production of sestrins. Their effects are associated with their antioxidant defense ability and regulation of intracellular signaling pathways, such as double direction regulating and controlling of the target of rapamycin kinase complexes 1 and 2 (mTORC1 and mTORC2), regulating autophagy, and activating the AMP-activated protein kinase (AMPK) pathway.16, 17 Furthermore, because of their high sensitivity to stress, sestrins can quickly respond to various damages, protecting the organism from stress. Recently, studies have reported Sesn1 to play a significant role in cardiovascular and cerebrovascular diseases by reducing neuronal injury caused by oxygen-glucose deprivation/reoxygenation in vitro and doxorubicin cardiotoxicity in vivo.14, 18 However, whether Sesn1 alleviated myocardial IRI was unknown. In addition, Sesn1 was regulated by p53, a key gene in ferroptosis.19, 20 Therefore, we suspected Sesn1 to play a crucial cytoprotective role in ferroptosis involving myocardial IRI.
Although some studies have indicated that ferroptosis is involved in myocardial injury caused by AMI, we do not know whether it plays a role in the ischemia phase or the reperfusion phase. Furthermore, extensive studies have verified the role of Sesn1 in cell survival and oxidative stress under various conditions, but little is known about whether Sesn1 is involved in ferroptosis regulating myocardial IRI. Therefore, experimental research is required to clarify this matter.
Objectives
This study aimed to simulate clinical AMI occurring within 6 h in rats, investigate the differences in ferroptosis between the phases of ischemia and reperfusion, and further explore the relationship between ferroptosis, IRI and Sesn1.
Materials and methods
Animals
Adult male Sprague Dawley (SD) rats of specific pathogen-free (SPF) grade (250–280 g) were provided by the Animal Laboratory Center, Guangxi Medical University, Nanning, China. The rats were raised by professional breeders and their living environment was strictly controlled. The rats were adaptively fed for 3–5 days before the operation. The Institutional Animal Care and Research Ethics Committee of Guangxi Medical University approved the plan for the animal experiment (approval No. 2022-KY-E-(227)). All procedures complied with the regulations specified by the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Ischemia/reperfusion injury model
The rats were anesthetized by intraperitoneal sodium pentobarbital (40 mg/kg) injection and fixed on the operating table in the supine position. They were intubated through the larynx, and the endotracheal tube was connected to an animal ventilator to assist breathing. The mode of the ventilator was 80 bpm in respiratory rate, 20 mL/kg in tidal volume, and a respiratory ratio of 1:1.5. The myocardial infarction model was created by ligating the left anterior descending coronary artery (LAD) with a 6-0 silk suture.
The first part of the experiments was to confirm whether ferroptosis was involved in ischemia-induced myocardial injury. To simulate clinical AMI presenting within 6 h, the animals were randomly divided into 4 groups (n = 6 per group), including sham group (only threading without ligation), ischemia 2-hour group, ischemia 4-hour group, and ischemia 6-hour group. After the operation was finished, blood was collected and centrifuged, and serum was taken for the detection of creatine kinase-MB (CK-MB) and malondialdehyde (MDA). In addition, the myocardium was removed from the rats to measure the expression of glutathione peroxidase 4 (GPX4), ferritin heavy chain 1 (FTH1) and iron content.
The purpose of the 2nd part of the experiments was to observe the occurrence and changes of ferroptosis in hearts of rats undergoing different reperfusion periods after suffering ischemia. Ninety rats were randomly divided into 3 clusters (n = 30 per cluster), and each cluster was subsequently divided into 5 groups (n = 6 per group). In briefly, the 1st cluster included the sham group, the ischemia 2-hour plus reperfusion groups (reperfusion 3-hour (R3h) group, reperfusion 6-hour (R6h) group, reperfusion 12-hour (R12h) group, and reperfusion 24-hour (R24h) group); the 2nd cluster was divided into the sham group and ischemia 4-hour plus reperfusion groups (R3h group, R6h group, R12h group, and R24h group); the 3rd cluster consisted of the sham group and the ischemia 6-hour plus reperfusion groups (R3h group, R6h group, R12h group, and R24h group). After 2 h, 4 h or 6 h of myocardial ischemia, the ligation wires were loosened and reperfusion was performed for another 3 h, 6 h, 12 h, or 24 h, respectively. When the experiment was over, serum and myocardial tissue were saved to detect the indexes of CK-MB, MDA, GPX4, and FTH1, as well as iron content. The time points of 2 h of ischemia plus 12 h of reperfusion were selected for the 3rd part of the experiments.
The 3rd part of the experiments evaluated the effects of ferroptosis on myocardial IRI. The time points of 2 h of ischemia plus 12 h of reperfusion were selected for the 3rd part of the experiments. It was a new cluster consisting of 3 groups (the sham group, the ischemia/reperfusion (I/R) group and the + Fer-1 group). Rats were treated with Fer-1 (3 mg/kg, intraperitoneal injection) before reperfusion. Finally, the serum of the rats was taken for CK-MB and MDA detection, and the myocardium was removed to measure the expression of GPX4, FTH1 and Sesn1 as well as iron content, to calculate the myocardial infarction area, and to detect the histopathological injury. The morphological changes of the mitochondria in each group were observed using transmission electron microscope (TEM).
Measurements of serum biochemistry
and iron content
Creatine kinase-MB is a creatine kinase isoenzyme found primarily in heart tissue. An increase in CK-MB indicates myocardial cell injury or necrosis. Malondialdehyde is the stable metabolite of lipid peroxidation products that indirectly reflects the degree of myocardial cell damage. Iron content is a common biomarker of ferroptosis. The serum MDA level in the myocardial tissues was measured using an MDA assay (No. A003-1; Nanjing Jiancheng Bioengineering Institute, Nanjing, China), serum CK-MB level was measured using a CK-MB assay (No. C060; Changchun Huili Biotech Co., Ltd., Changchun, China) and iron content was measured using a iron content assay (Solarbio, Beijing, China), all according to manufacturers’ instructions.
Histological observation
Hematoxylin and eosin (H&E) staining detected histopathological injury to the myocardium. After the heart was isolated, the tissue was fixed, embedded, sliced, dewaxed, and stained. The sections were then observed under a light microscope (Olympus BX53F; Olympus Corp., Tokyo, Japan).
Infarct area assessment
The assessment of the infarct area directly revealed the death of cardiomyocytes. The myocardial infarction area was calculated by staining the heart slice with 2% 2,3,5-triphenyltetrazolium chloride (TTC; Solarbio), according to the manufacturer’s instructions. The judgment criteria were that the normal tissue remained red and the infarct area was pale. The infarct area of the processed cardiac tissue section was calculated and analyzed using ImageJ software (National Institutes of Health, Bethesda, USA). The infarct area was expressed as the ratio of the infarction area to the total myocardial area.
Western blot
Both GPX4 and FTH1 are well-recognized biomarkers of ferroptosis. Focusing on Sesn1 was one of the key objectives during the 3rd experiment. Therefore, western blot was used to detect the difference of GPX4, FTH1 and Sesn1 protein levels among groups. It was carried out using the standard protocol. First, the myocardial tissue was lysed with radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors (Solarbio). Next, the total protein concentration was determined using the BCA protein assay kit (Beyotime, Nanjing, China) and the supernatant from centrifugation. Next, an equal quantity of protein samples was used to perform electrophoresis (10% or 7.5% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE)), membrane transfer (0.22 µm polyvinylidene difluoride (PVDF) membranes), blocking (5% skim milk), and incubation with primary antibodies, such as GPX4 (No. 67763-1-Ig; Proteintech, Rosemont, USA), FTH1 (No. A19544; Abclonal, Woburn, USA) and Sesn1 (No. ab134091; Abcam, Cambridge, UK), in a 4°C refrigerator overnight, and was finally treated with secondary antibodies (No. SA5-35521 and No. SA5-35571; Invitrogen, Waltham, USA). The endogenous control was α-tubulin (No. 66031-1-Ig; Proteintech). The results were visualized and analyzed using ImageJ software. The results of the targeting proteins were normalized against control, and the results were expressed as a fold of the control.
Quantitative real-time polymerase
chain reaction assay
Quantitative real-time polymerase chain reaction assay (qRT-PCR) was carried out to detect the gene expression of GPX4, FTH1 and Sesn1 according to the standard protocol. The TriZol reagent (No. 9108; TaKaRa Bio Inc., Kusatsu, Japan) was used to collect the total RNA. The cDNA was synthesized from 1 µg of total RNA using MonScriptTM RTIII All-in-one Mix Kit (No. MR05101; Monad Biotech, Wuhan, China), according to the manufacturer’s protocol. Subsequently, qRT-PCR was performed using the MonAmpTM SYBR Green qPCR Mix Kit (No. MQ10301S; Monad Biotech) on Applied Biosystems 7500 Real-Time PCR Systems (Thermo Fisher Scientific, Waltham, USA). The total reaction volume was 20 µL per well. The amplification protocol comprised of the following PCR cycles: a single cycle of 30 s at 95°C, 40 cycles of 10 s at 95°C, a single cycle of 30 s at 60°C, and a final cycle of 15 s at 95°C. The expression levels of β-actin normalized the threshold cycle values for target genes. The relative gene expression data were calculated using the 2−ΔΔCT method. The primers (Sangon Biotech, Shanghai China) were as follows:
1. β-actin: forward (5’ > 3’) TGTCACCAAACTGGGACGATA; reverse (5’ > 3’) GGGGTGTTGAAGGTCTCAAA.
2. GPX4: forward (5’ > 3’) GATACGCCGAGTGTGGTTT; reverse (5’ > 3’) CTTGGGCTGGACTTTCATC.
3. FTH1: forward (5’ > 3’) GCCAAATACTTTCTCCATCAA; reverse (5’ > 3’) TCATCACGGTCAGGTTTCT.
4. Sesn1: forward (5’ > 3’) ACCTCGTGACCCTGACTTT; reverse (5’ > 3’) GCTCATTTACCCCAAACCT.
TEM observation
The mitochondrial reduction is one of the characteristics of ferroptosis. Therefore, TEM was selected to observe the morphological changes of the myocardial mitochondria. At the end of the experiment, the rats were euthanized by excessive anesthesia, and their hearts were removed. Then, a 1-mm3 piece was cut from the cardiac apex and fixed in an electron microscope fixative solution. After preparing the specimens, the samples were observed and imaged with a TEM system (model HT7800; Hitachi, Tokyo, Japan).
Statistical analyses
The IBM SPSS v. 23.0 software (IBM Corp., Armonk, USA) was used to perform statistical analysis. Intergroup data having independent, normal distribution and variance homogeneity were analyzed using one-way analysis of variance (ANOVA), followed by Tukey’s honestly significant difference (HSD) test. If the data did not meet these conditions, the differences between groups were determined using the nonparametric Kruskal–Wallis test, followed by Dunn’s post hoc test. If the data had normal distribution and homogeneity of variance, independent sample t-tests were performed between the groups. A p-value <0.05 indicated statistical significance.
Results
Ferroptosis did not occur during myocardial ischemia in rats
To verify whether ferroptosis was involved in myocardial ischemia injuries, rat hearts were subjected to ischemia for 2 h, 4 h and 6 h, respectively. Ischemia led to myocardial injury in rats and such injury was aggravated by prolonged periods of ischemia, manifested as a gradual increase in CK-MB (p < 0.001; Figure 1A). These results suggested that the myocardial ischemia injury model was established successfully. Although ferroptosis was iron-dependent, there were no differences in iron content in myocardial tissues between the sham group and ischemia groups (sham group compared to ischemia 2-hour group: p = 0.992; compared to ischemia 4-hour group: p = 0.427; compared to ischemia 6-hour group: p = 0.834; Figure 1B). Similarly, compared with the sham group, the concentration lipid peroxidation product, MDA, was not significantly higher in the ischemia groups (sham group compared to ischemia 2-hour group: p = 0.083; compared to ischemia 4-hour group: p = 0.756; compared to ischemia 6-hour group: p = 0.085; Figure 1B). There were no significant changes in protein expression of GPX4 and FTH1, well-recognized biomarkers of ferroptosis, between the sham group and ischemia groups (GPX4: sham group compared to ischemia 2-hour group: p = 0.808; compared to ischemia 4-hour group: p = 0.894; compared to ischemia 6-hour group: p = 0.994; FTH1: sham group compared to ischemia 2-hour group: p = 0.957; compared to ischemia 4-hour group: p = 0.979; compared to ischemia 6-hour group: p = 0.988; Figure 1C,D). Based on the above evidence, we concluded that ferroptosis did not occur during myocardial ischemia.
Ferroptosis participated in myocardial IRI
This part of the research aimed to observe the changes of ferroptosis in the hearts of rats that underwent ischemia for different times (2 h, 4 h and 6 h) and different reperfusion periods (R3 h, R6 h, R12 h, and R24 h). In the ischemia 2-hour plus reperfusion groups, CK-MB level (Figure 2A) increased in the reperfusion groups (compared to the sham group: p < 0.001), and peaked at R12h (compared to R3h: p < 0.001; compared to R6h: p = 0.008; compared to R24h: p < 0.001). As illustrated in Figure 2B, there was a significant difference in MDA level between the sham group and reperfusion groups (compared to the sham group: for R3h p = 0.006, for R6h, R12h and R24h p < 0.001). The peak formed at R12h (compared to the R3h: p = 0.049; compared to R6h: p = 0.421; compared to R24h: p = 0.538). The iron content (Figure 2B) was significantly different between the sham group and the reperfusion groups (p < 0.001) and there were no statistical differences between reperfusion groups. In addition, as shown in Figure 2C,D, the protein levels of GPX4 and FTH1 decreased in the reperfusion groups; the decreases began 3 h after myocardial reperfusion (compared to sham group: p < 0.001). There was no significant difference in GPX4 protein level along with myocardial reperfusion prolongation. An upward trend of FTH1 could be observed at R3h (R3h compared to R12h: p = 0.025; R3h compared to R24h: p = 0.003).
As demonstrated in Figure 3A, in the ischemia 4-hour plus reperfusion groups, CK-MB level increased in the reperfusion groups (compared to sham group: p < 0.001) and peaked at R12h (compared to R3h and R6h: p < 0.001; compared to R24h: p = 0.172). Compared to the sham group, both MDA level and iron content (Figure 3B) increased in the reperfusion groups (MDA: for R12h p = 0.008, for R24h p = 0.022; iron content: for R3h, R6h R12h, and R24h p < 0.001). There were no statistical differences between the reperfusion groups. Compared to the sham group, the protein levels of GPX4 and FTH1 (Figure 3C,D) decreased significantly, which began at R3h (for GPX4: p = 0.023, for FTH1: p < 0.001). No significant differences in GPX4 and FTH1 were found between the reperfusion groups.
In the ischemia 6-hour plus reperfusion groups, compared with the sham group, CK-MB level (Figure 4A) also increased in the reperfusion groups (p < 0.001) and peaked at R24h (compared to R3h and R6h: p < 0.001; compared to R12h: p = 0.004). As shown in Figure 4B, an upward trend of MDA level can be observed among the sham group and reperfusion groups (compared to the sham group: for R3h p = 0.12, for R6h p = 0.048, for R12h p = 0.005, for R24h p < 0.001). However, there were no statistical differences between the reperfusion groups. Compared to the sham group, the iron content (Figure 4B) increased in reperfusion groups (for R3h: p = 0.001; for R6h, R12h and R24h: p < 0.001) and peaked at R6h (compared to R3h: p < 0.001; compared to R12h and R24h: p = 1.000). There were significant differences in the protein expression of GPX4 and FTH1 (Figure 4C,D) between the sham group and reperfusion groups. The decrease of GPX4 and FTH1 began at R6h (p < 0.001). There was no significant difference in GPX4 along with myocardial reperfusion prolongation. An upward trend of FTH1 was observed in the R6h group (R6h compared to R12h: p = 0.015; R6h compared to R24h: p < 0.001; R12h compared to R24h: p < 0.001).
Administration of Fer-1 alleviated myocardial IRI in rats
The Fer-1, a ferroptosis inhibitor, was intraperitoneally injected into rats before reperfusion to evaluate the contribution of ferroptosis to myocardial IRI. Compared to the I/R group, Fer-1 significantly reduced myocardial IRI in the +Fer-1 group. The rise of serum CK-MB activity reversed (p < 0.001; Figure 5A), the area of myocardial infarction decreased (p = 0.008; Figure 5B,C) and the injury of myocardial pathological tissue was alleviated (Figure 5D). The results verified that inhibition of ferroptosis played a therapeutic role in myocardial IRI.
Treatment with Fer-1 alleviated myocardial IRI in an iron-dependent manner and influenced the expression of Sesn1
The levels of MDA, iron content, GPX4, FTH1, Sesn1, and mitochondrial ultrastructure in the myocardium were all measured simultaneously to detect the effects of Fer-1. Compared to the sham group, MDA level and iron content (Figure 6A) increased in the I/R group (for MDA: p < 0.001; for iron content: p = 0.004). The increased effect of MDA induced by I/R can be significantly blocked by Fer-1 (for I/R group compared to +Fer-1 group: p = 0.008); however, Fer-1 had no effect on the iron content (for I/R group compared to +Fer-1 group: p = 0.589). As shown in Figure 6B, the transcription level of GPX4, FTH1 and Sesn1 decreased in the I/R group (compared to the sham group: for GPX4 p < 0.001, for FTH1 p = 0.001 and for Sesn1 p < 0.001); in addition, treating with Fer-1 can reduce the decline of Sesn1, but not GPX4 and FTH1 (I/R group compared to +Fer-1 group: for GPX4 p = 0.142, for FTH1 p = 0.557 and for Sesn1 p = 0.025). The trends for GPX4, FTH1 and Sesn1 protein expression levels were consistent with the transcription levels. As demonstrated in Figure 6C–E, owing to the occurrence of ferroptosis during myocardial I/R in rats, the protein expression level of GPX4, FTH1 and Sesn1 decreased (compared to the sham group: p < 0.001); however, this influence can be reversed by Fer-1 (I/R group compared to +Fer-1 group: for GPX4 and FTH1 p < 0.001, for Sesn1 p = 0.017). In addition, characteristic mitochondrial changes, such as smaller mitochondria, increased membrane density and decreased or fractured cristae, occurred in the I/R group due to ferroptosis. At the same time, Fer-1 alleviated these mitochondrial changes (Figure 6F). In summary, the treatment with Fer-1 can alleviate myocardial IRI in an iron-dependent manner. Sestrin 1 was differentially expressed in ferroptosis involving myocardial IRI model.
Discussion
In this research, our primary objective was to explore whether ferroptosis is involved in myocardial IRI in vivo. Our findings were as follows: 1) Ferroptosis was not observed in rat hearts that underwent ischemia from 2 h to 6 h, although the ischemic injury worsened with prolonged ischemia. 2) Ferroptosis occurred in rat hearts that suffered ischemia for 2 h, 4 h or 6 h plus reperfusion from 3 h to 24 h, and the duration of ischemia determined the appearance time of ferroptosis during myocardial reperfusion. 3) The administration of Fer-1 significantly attenuated myocardial IRI by inhibiting ferroptosis. 4) Treating rats with Fer-1 reversed the decrease in the expression of Sesn1 caused by myocardial IRI. This study demonstrated the role ferroptosis plays in the ischemic and reperfusion phases of myocardial IRI. In addition, this study proposed that Sesn1 was associated with ferroptosis in myocardial IRI.
It is without a doubt that ischemia can lead to myocardial injury, and the fact that reperfusion causes injury cannot be ignored. The study was initially designed based on the following considerations: in clinical practice, it can be observed that some patients refuse PCI for personal reasons or are not suitable candidates for PCI. In these cases, limited effects of other treatment result in permanent myocardial injury. On the other hand, with growing awareness of AMI, more patients see a doctor in time to open the infarct-related arteries. However, further aggravation of myocardial injury occurs following reperfusion. Although many studies have tried to explain the mechanisms of myocardial ischemia injury and reperfusion injury, the difference between them has not been fully clarified, and the effects of clinical therapies have been disappointing. The current research hotspot, ferroptosis, has aroused our interest to contribute to the distinction between myocardial ischemia injury and reperfusion injury, laying a basis for precision medicine.
Next, we would like to discuss ischemic time and why we chose persistent ischemia for 2 h, 4 h and 6 h. At present, the ligation of the LAD is a mature model of AMI in animals.21 According to most studies, the ischemic time of AMI models is less than 1 h. The rodent heart is in a state of reversible injury when the myocardial ischemia time is too short and will not cause apparent infarction. However, once the ischemia time is too long, transmural infarction occurs, which can induce the death of model animals during the experiment. In the pre-experiment phase, we found that rats can tolerate myocardial ischemia well. The longest ischemia time was 1 week, and still the mortality of these rats was low. It was found that ferroptosis occurred distinctly in rats after 1 h of myocardial ischemia followed by reperfusion. However, no differences were observed in iron and MDA content between the reperfusion and ischemia groups, except for a visible, increasing trend in the reperfusion group.22 The researchers explained that ferroptosis might occur slightly in rats with cardiac ischemia. Since the time of myocardial ischemia they chose was less than 1 h, we wondered whether the apparent ferroptosis could be observed by prolonging the time of ischemia.
On the other hand, the optimum treatment time for myocardial reperfusion is within 6 h. Therefore, this study aimed to simulate clinical AMI within 6 h using a rat model. Significant differences exist between humans and animals. Although we do not know the ideal treatment time of AMI in rats, our purpose was to explore whether ferroptosis was involved in myocardial injury resulting from ischemia within or beyond the treatment time window or from reperfusion, which would contribute to precision medicine and save the damaged cardiomyocytes to the greatest extent. Therefore, we designed 3 time points (2 h, 4 h and 6 h) to dynamically observe the change during myocardial infarction. In the 2nd experiment, 4 reperfusion subgroups according to the reperfusion time (R3h, R6h, R12h, and R24h) were constructed. Our design aimed to observe the changes in ferroptosis during the extension of reperfusion.
Iron participates in various metabolic processes, such as electron transport, DNA synthesis and oxygen transport. Several studies have shown ferroptosis to be an important form of cell death in cardiomyocytes.23, 24, 25 Evidence demonstrates that ferroptosis occurs primarily due to the lethal accumulation of lipid peroxidation products, which are iron-dependent due to glutathione (GSH) depletion or inhibition of system Xc− (a cystine/glutamate antiporter system).26 Several vital metabolic pathways are involved in this process. The 1st is the iron metabolic pathway – iron overload produces lipid peroxidation products through the Fenton reaction. The 2nd is GSH production and the GPX4 pathway, which plays a protective role to reduce lipid peroxidation. The 3rd is the glucose metabolism, which generates nicotinamide adenine dinucleotide phosphate (NADPH) through pentose phosphate pathways. NADPH, as the coenzyme of GSH, plays an important role in maintaining the content of GSH in cells.23 Ferroptosis was reported to be related to the pathogenesis of many diseases, such as cancer, neurological diseases and sepsis.27, 28, 29 Recent studies found that ferroptosis is involved in myocardial IRI. The levels of iron, MDA and the expression of ACSL4 in reperfused rat hearts were gradually increased, with a decreased GPX4 level compared to the control group.22 Similarly, in our study, we found that ferroptosis was stimulated in the myocardial I/R group, which was evidenced by the decreased expression of GPX4 and FTH1, and the increased levels of iron and MDA. In particular, we were surprised to find that the duration of ischemia determined the appearance time of ferroptosis during myocardial reperfusion. In the myocardial ischemia (2 h and 4 h) plus reperfusion groups, ferroptosis biomarkers changed significantly at R3h, which meant that ferroptosis occurred in rat hearts. However, when the myocardial ischemia was prolonged to 6 h, ferroptosis emerged later – at R6h. In our opinion, this result can be explained as follows: Since ferroptosis only occurs during the myocardial reperfusion phase rather than the ischemia phase, ferroptosis was found to be closely related to blood flow. The longer the coronary artery occlusion, the more serious the damage to the vascular structure and function. The total time of myocardial ischemia was frequently reported to be associated with an increased incidence of the no-reflow phenomenon.30, 31 Research revealed that prolonged total ischemic time was an independent predictor of the no-reflow phenomenon.32 The present study depicted that no matter how long the time of myocardial ischemia (2–6 h), ferroptosis only occurred during the reperfusion phase, and the myocardial injury was alleviated after the administration of a ferroptosis inhibitor – Fer-1. Our research provided a definite time orientation for ferroptosis in myocardial IRI and a basis for clinical treatment.
Sestrin 1 is essential for maintaining normal function during oxidative injury. Its overexpression suppresses inflammation and apoptosis induced by oxidized low-density lipoproteins (ox-LDL) in human umbilical vein endothelial cells.33 Silencing Sesn1 can guide the inflammation and lipid accumulation in RAW264.7 cells exposed to ox-LDL.34 Research showed that after being fed a Western diet for 8 weeks, Sesn1 knockout mice exhibited higher levels of hepatic lipotoxicity and oxidative stress than their wild-type counterparts.15 These findings suggested an antioxidant function of Sesn1. Over the past few years, extensive research has revealed that the essence of ferroptosis is an iron-dependent accumulation of lipid peroxidation induced by oxidative stress.35, 36, 37, 38 Recently, researchers have verified that the interactions between Sesn1, p53 and nrf2 participate in many pathological processes.14, 39, 40, 41 While p53 and nrf2 are the hub genes for ferroptosis, we speculated that there was a connection between Sesn1 and ferroptosis. Oxidative stress was one of the crucial mechanisms of myocardial IRI. However, the relationship between Sesn1, ferroptosis and myocardial IRI was unclear. In our study, the expression of Sesn1 in rat hearts decreased significantly after ischemia (2 h)/reperfusion (12 h), but this effect was reversed after the administration of Fer-1. In addition, we confirmed that the inhibition of ferroptosis can alleviate myocardial IRI. In summary, Sesn1 is involved in the mechanism by which ferroptosis regulates myocardial IRI.
Limitations
There were some limitations of this study that should be put forward for discussion to guide follow-up research. First, whether the inhibition of ferroptosis can protect against myocardial ischemia is unknown; however, there were no signs of ferroptosis in the ischemia group. Second, in this study, we verified that Sesn1 was differentially expressed in myocardial IRI. However, we failed to confirm the effects of its overexpression or knockdown on myocardial IRI. Lastly, the upstream and downstream pathways of Sesn1 involved in myocardial IRI were not elucidated. However, these limitations will help direct future research and our further studies.
Conclusions
Our findings verified that ferroptosis occurred in the phase of myocardial reperfusion but not ischemia. We are the first to report that a longer duration of myocardial ischemia resulted in a later occurrence of ferroptosis during reperfusion. Moreover, the inhibition of ferroptosis alleviated reperfusion-induced myocardial damage. Our findings provide a theoretical basis for specific interventions targeting ferroptosis in AMI. Furthermore, we discovered Sesn1 to be involved in myocardial IRI, laying the groundwork for future research.





