Abstract
Background. Diabetic nephropathy (DN) is one of the most common complications of diabetes mellitus (DM). MicroRNA (miR)-218 is associated with the development of diabetes. Besides, sprouty-related EVH1 domain containing 2 (SPRED2), the downstream target of miR-218, is involved in insulin resistance and inflammation.
Objectives. Since inflammation plays a key role in DN, and SPRED2 is known to facilitate cell autophagy, the present study aimed to investigate the role and molecular mechanism of miR-218 and SPRED2-mediated autophagy in high glucose (HG)-induced renal tubular epithelial cells using an in vitro model.
Materials and methods. The HK-2 cells were cultured in 5.5 mM or 30 mM D-glucose medium. Quantitative real-time polymerase chain reaction (qRT-PCR) was used to detect the expression of miR-218 and SPRED2. Western blotting was performed to calculate the levels of SPRED2, inflammatory cytokines, autophagy-related and apoptosis-related proteins. Reactive oxygen species (ROS) level was evaluated using cellular ROS assay kit, superoxide dismutase (SOD) activity was detected using SOD activity assay kit, and malondialdehyde (MDA) content was measured using lipid peroxidation. The levels of interleukin (IL)-1β, IL-6, IL-4, and tumor necrosis factor alpha (TNF-α) were detected with enzyme-linked immunosorbent assay (ELISA). Cell apoptosis was evaluated using flow cytometry analysis. The targeting relationship between miR-218 and SPRED2 was identified with a luciferase reporter. The LC3-II expression was detected with immunofluorescence.
Results. The miR-218 expression was upregulated and SPRED2 expression was downregulated in HG-induced HK-2 cells. The miR-218 was proven to target SPRED2 and negatively regulate SPRED2 expression. Besides, downregulated miR-218 alleviated inflammatory response, oxidative stress and cell apoptosis, but aggravated autophagy. We also showed that downregulated SPRED2 reversed the effect of miR-218 on inflammation, cell apoptosis and autophagy in HG-induced HK-2 cells.
Conclusions. The miR-218 can promote oxidative stress and inflammatory response in HG-induced renal tubular epithelial cells by inhibiting SPRED2-mediated autophagy. This study might bring novel understanding for molecular mechanism of DN.
Key words: miR-218, renal tubular epithelial cell, autophagy, SPRED2, high glucose
Background
Diabetic nephropathy (DN) is one of the most common complications of diabetes mellitus (DM) that might eventually develop into chronic nephropathy.1, 2, 3 During the development of diabetes, high glucose (HG) condition can induce cell apoptosis, inflammation and oxidative stress in renal cells, e.g., renal tubular epithelial cells, leading to renal injuries, and finally resulting in DN and chronic nephropathy.4, 5, 6 Therefore, it is of great importance to find new and potential biomarkers involved in renal tubular cells for better DN therapy.
Currently, various kinds of miRNAs have been illustrated to play important roles in diabetes and its complications by regulating biological pathways related to DN.7, 8, 9 As reported, kidney hypoxia triggered the upregulation of miR-218 expression in endothelial progenitor cells that might promote endocapillary repair.10, 11 The miR-218 was also found to be a biomarker for type 1 DM in children.12 Moreover, as stated in the study by Zhang et al., miR-218 was upregulated in the plasma samples of patients with type 2 DM-induced atherosclerosis.13 Circulating evidence indicated that miR-218 plays a mediatory role in the pathogenesis of DN. However, deeper insight is still needed to illustrate the role of miR-218 in the development of DN.
As a member of sprouty-related EVH1 domain containing proteins family, sprouty-related EVH1 domain containing 2 (SPRED2) is usually expressed in various tissues.14, 15, 16 It has been reported that SPRED2 negatively regulated high-fat diet-induced obesity, adipose tissue inflammation, metabolic abnormalities, and insulin resistance.17 The SPRED2 was identified as a novel regulator of cardiac autophagy, and its deficiency could arouse cardiac dysfunction and life-threatening arrhythmias through impaired autophagy.18, 19, 20 In addition, SPRED2 affected the development of lipopolysaccharide-induced lung inflammation by negatively regulating the ERK-MAPK pathway.21 Since SPRED2 influences diabetes-related diseases including insulin resistance, obesity, as well as inflammation, which plays a key role in DN, it may also participate in DN process. However, limited research revealed the role of SPRED2 in autophagy, inflammation and oxidative stress during DN development.
Objectives
In the present study, we investigated the molecular mechanism of miR-218 and SPRED2 and their actions in DN in a well-accepted in vitro model using HG-induced HK-2 cells. Many studies used similar in vitro models to investigate DN.22, 23, 24 These findings might provide new promising therapeutic strategies for DN.
Materials and methods
Cell culture and treatment
Renal tubular epithelial cells (HK-2) were purchased from American Type Culture Collection (ATCC; Rockville, USA). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies BRL, Gaithersburg, USA) supplemented with fetal bovine serum (FBS), 100 μg/mL streptomycin and 100 U/mL penicillin (Gibco BRL, Carlsbad, USA) at 37°C with 5% CO2 in appropriate humidity.
The HK-2 cells were divided into a HG group and a normal glucose (NG) group. The HK-2 cells in the HG group were treated with a HG concentration of 30 mM, while the NG cells were treated with 5.5 mM glucose. Both groups were incubated with 5% CO2 at 37°C for 48 h. For inhibition of cell autophagy, 3-MA (Sigma-Aldrich, St. Louis, USA) was used to treat the cells with a dose of 3 mmol/L.
Cell transfection
The cells were transfected with 5 nM miR-218 mimics/inhibitor, as well as si-SPRED2, and the corresponding negative controls (NCs). The miRNA mimics, inhibitor and NCs were purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China) without sequence information. The si-SPRED2 was purchased from Sigma-Aldrich (cat. No. EHU029561) with sequence of TCCATGGTGAACGACAGAAAGACAAACTGGTGGTATTGGAATGCTATGTAAGAAAGGACTTGGTCTACACCAAAGCCAATCCAACGTTTCATCACTGGAAGGTCGATAATAGGAAGTTTGGACTTACTTTCCAAAGCCCTGCTGATGCCCGAGCCTTTGACAGGGGAGTAAGGAAAGCAATCGAAGACCTTATAGAAGGTTCAACAACGTCATCTTCCACCATCCATAATGAAGCTGAGCTTGGCGATGATGACGTTTTTACAACAGCTACAGACAGTTCTTCTAATTCCTCTCAGAAGAGAGAGCAACCTACTCGGACAATCTCCTCTCCCACATCCTGTGAGCACCGGAGGATTTATACCCTGGGCCACCTCCACGACTCATACCCCACAGACCACTATCACCTCGATCAGCCG. The siRNA negative control was also obtained from Sigma-Aldrich (cat. No. SIC001) with meaningless sequence. Cell transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, USA) and detection of transfection efficiency was conducted at 48 h.
Flow cytometry analysis
The treated HK-2 cells were collected into 1.5 mL tubes containing annexin-FITC and propidium iodide (PI) reagents, and cultured in the dark at room temperature for 20 min. Afterwards, 200 μL of PI reagents, with 1 mL phosphate-buffered saline (PBS) were added into the flow tube. Cell apoptosis was quantified using FACSCalibur (Becton Dickinson, Mountain View, USA).
ELISA
The cell supernatant expression of tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, IL-4, and IL-6 was determined with enzyme-linked immunosorbent assay (ELISA) using commercially available kits (Human TNF alpha ELISA Kit (ab181421), Human IL-1 beta ELISA Kit (ab214025) and Human IL-4 ELISA Kit (ab215089), all purchased from Abcam, Cambridge, USA) according to the manufacturer’s instructions.
Measurement of superoxide dismutase, malondialdehyde and reactive oxygen species generation
Reactive oxygen species (ROS) level was evaluated using cellular ROS assay kit (Deep Red; ab186029), superoxide dismutase (SOD) activity was detected by means of superoxide dismutase activity assay kit (Colorimetric; ab65354), and malondialdehyde (MDA) content was measured using Lipid Peroxidation (MDA) Assay Kit (Colorimetric/Fluorometric; ab118970), all purchased from Abcam.
qRT-PCR
Total RNA was extracted from HK-2 cells using Trizol reagent (Invitrogen, Carlsbad, USA). For the detection of miR-218 expression, a miRcute miRNA First-strand cDNA synthesis kit (Tiangen Biotech Co., Beijing, China)was used to convert RNA into cDNA. A miRcute miRNA qPCR detection kit (Tiangen Biotech Co.), performing on a 7900 HT Sequence Detection System (Applied Biosystems, Foster City, USA) was used in quantitative real-time polymerase chain reaction (qRT-PCR). For the detection of SPRED2 expression, a PrimeScript RT reagent Kit (TaKaRa, Tokyo, Japan) was used to convert RNA into cDNA. A Power SYBR Green kit (Thermo Fisher Scientific) with the Stratagene Mx3000P real-time PCR system (Stratagene, La Jolla, USA) was conducted for quantification. The following primers were used in PCRs: F 5’-TGTGAGCACCGGAAGATTTATACC-3’ and R 5’-CGCGGCGGCTTTGTGCTT-3’ for SPRED2; F 5’-TAATGGTCGAACGCCTAACGTC-3’ and R 5’-CGAGTGCATTTGTGCTTGATCTA-3’ for miR-218; 5’-GACACGCAAATTCGTG-3’ and 5’-GTGCAGGGTCCGAGGT-3’ for U6, and F 5’-GGGTGTGAACCACGAGAAAT and R 5’-ACTGTGGTCATGAGCCCTTC-3’ for GAPDH. The U6 and GAPDH were used as internal controls for miRNA and mRNA, respectively. The relative expression level was calculated using the 2−ΔΔCq method.25
Western blotting
The protein extracted from HK-2 cells were loaded on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes blocked with 5% non-fat milk at a room temperature for 3 h. Afterwards, the membranes were incubated with primary antibody at 4°C overnight, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies at 37°C for 1 h. The primary antibodies were as follows: Anti-SPRED2 (ab153700, 1/500), Anti-caspase-3 (ab13847, 1/500), Anti-BAX (ab182733, 1/2000), Anti-BCL-2 (ab194583, 1/500), Anti-LC3-II/I (ab62721, 1 µg/mL), and Anti-Beclin 1 (ab207612, 1/2000) (all purchased from Abcam). The GAPDH was used as a control. Protein bands were detected using Super Signal West Pico Chemiluminescent Substrate kit (NCM Biotech, Suzhou, China).
Immunofluorescence
Briefly, cells were fixed in 4% paraformaldehyde for 5 min. For fluorescent labeling, cells were washed twice with PBS and permeabilized with 0.2% Triton X-100 for 5 min. Subsequently, cells were incubated with primary antibodies at 4°C overnight, followed with secondary antibodies at room temperature for 1 h. Fluorescence intensities were detected on an Olympus FluoView FV1000 confocal microscope (Olympus Corp., Tokyo, Japan).
Luciferase reporter assay
The binding mode between miR-218 and SPRED2 was predicted using TargetScan 7.2 (http://www.targetscan.org/vert_72/). The wild-type (WT) and mutation (MUT) of 3’-untranslated region (UTR) of SPRED2 sequences containing the binding sequence of miR-218 were amplified and inserted into the p-MIR-report plasmid (Ambion, Austin, USA). Afterwards, HK-2 cells were co-transfected with the miR-19b vector (inhibitor or mimics) or NCs in SPRED2-WT/SPRED2-MUTm, using Lipofectamine 3000 (Invitrogen). Cells were collected for detection using luciferase assay kits (Promega, Madison, USA) 48 h after the transfection. Luciferase activity of cells was normalized to Renilla luciferase activity.
Statistical analyses
The continuous data were presented as mean ± standard deviation (SD). The data normality in each compared group was evaluated with Shapiro–Wilk test. The comparison between 2 groups was made using the Student’s t-test, and the comparison among 3 or more groups was conducted using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. The Levene’s test was used for assumption of homogeneity of variances of ANOVA or t-test. The differences were considered statistically significant when p < 0.05. All calculations were performed using SPSS v. 18.0 (SPSS Inc., Chicago, USA).
Results
MiR-218 directly targets SPRED2 and negatively regulates SPRED2 expression
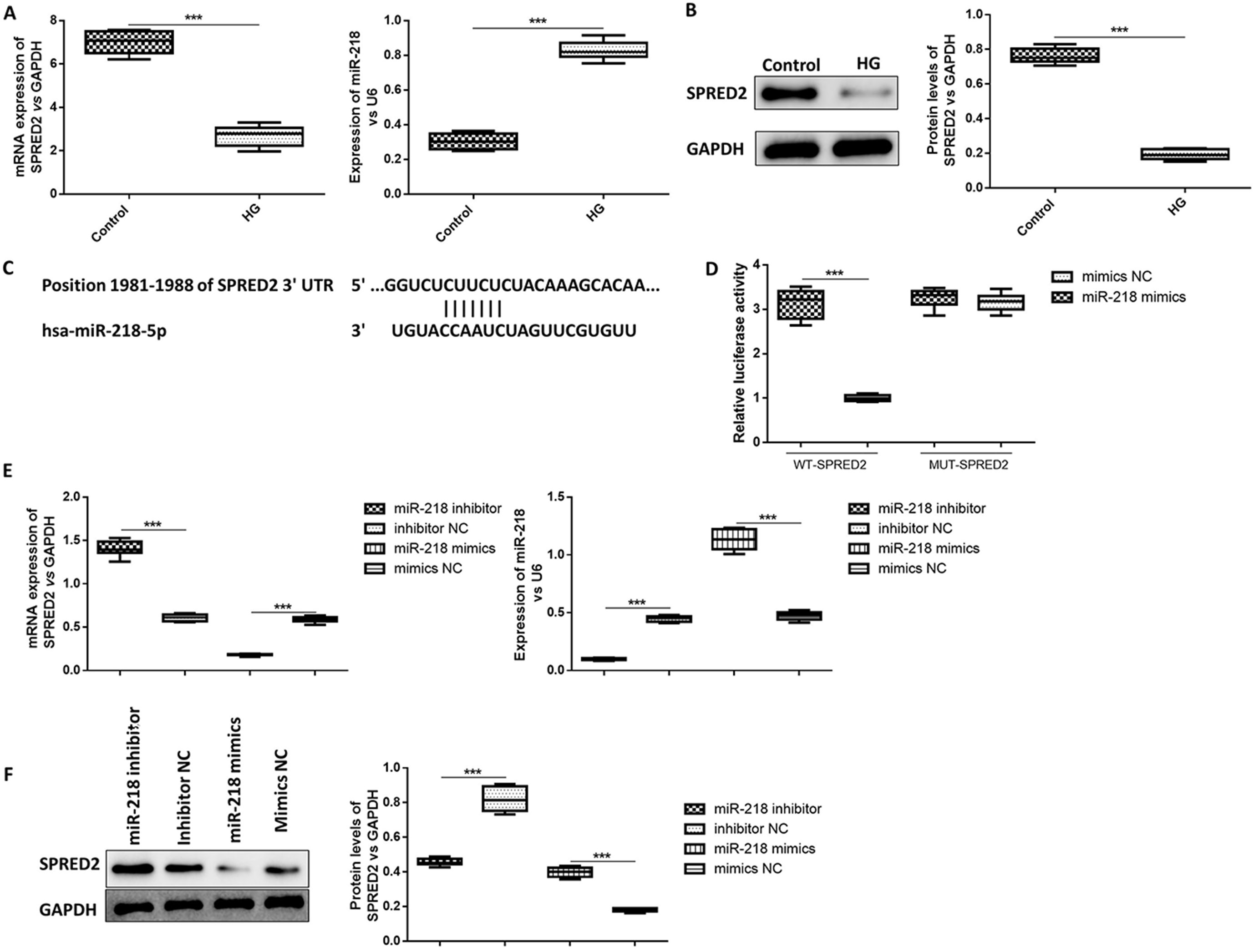
The expression levels of miR-218 and SPRED2 were detected under HG and NG conditions. As shown in Figure 1A,B, SPRED2 expression significantly decreased in HG-induced HK-2 cells compared to the NG group. Nevertheless, qRT-PCR result showed that miR-218 expression visibly increased in the HG group (Figure 1A). These findings suggested that miR-218 expression was upregulated and SPRED2 expression was downregulated in HK-2 cells under HG conditions.
To further investigate the molecular mechanism of miR-218 in HG-induced HK-2 cells, dual luciferase reporter assay was performed. Firstly, HK-2 cells were transfected with miR-218 mimics or miR-218 inhibitor, respectively, and the transfection efficiency was confirmed with qRT-PCR. Based on bioinformatics analysis, miR-218 was predicted to bind to the SPRED2 (Figure 1C). Luciferase reporter assay showed that the fluorescence intensity of SPRED2-WT was significantly decreased by the transfection of miR-218 mimics, but increased by the transfection of miR-218 inhibitor. However, miR-218 mimics/inhibitor had no effect on the fluorescence intensity of SPRED2-MUT (Figure 1D). As shown in Figure 1E, miR-218 expression was highly upregulated by the transfection of miR-218 mimics and knockdown by miR-218 inhibitor. Besides, both PCR and western blotting results proved that mRNA and protein levels of SPRED2 could be inhibited by overexpressed miR-218 but elevated by downregulated miR-218 (Figure 1E,F). The results of ANOVA test are listed in Table 1. The above results indicated that miR-218 directly targeted SPRED2 and negatively regulated SPRED2 expression.
Inhibition of SPRED2 or autophagy reverses the effect of miR-218 inhibitor on inflammation and oxidative stress in HG-induced HK-2 cells
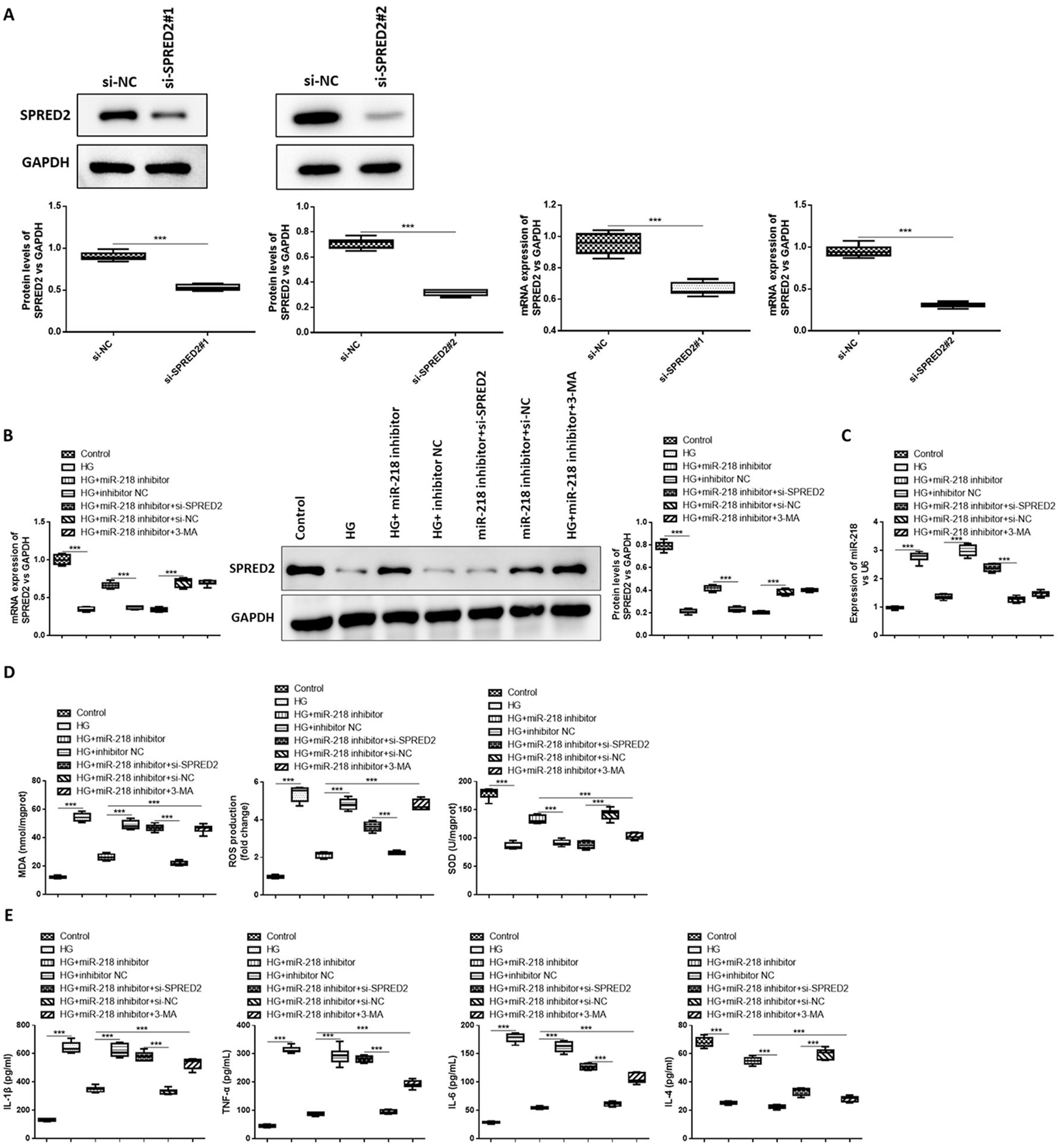
The study investigated further the effect of miR-218 and SPRED2 on inflammatory factor secretion and oxidative stress in HG-induced HK-2 cells. First, cells were transfected with si-SPRED2#1, si-SPRED2#2 or si-NC, respectively. The SPRED2 expression was remarkably downregulated by the transfection of si-SPRED2#1 or si-SPRED2#2, but si-SPRED2#2 showed better efficacy for knockdown of SPRED2 expression (Figure 2A). Hence, si-SPRED2#2 (abbreviated as si- SPRED2) was selected for the following experiments. Reduced SPRED2 expression induced by HG was elevated by miR-218 inhibitor; however, the co-transfection of si-SPRED2 reversed the effect of miR-218 inhibitor on SPRED2 expression (Figure 2B). Compared to the control, the levels of MDA and ROS were significantly increased and SOD expression was dramatically decreased in HG-induced HK-2 cells, but miR-218 inhibitor suppressed the effect of HG induction on HK-2 cells. At the same time, the transfection of si-SPRED2 or the treatment of 3-MA attenuated the effect of miR-218 inhibitor on oxidative stress factors (Figure 2C). Moreover, the increased levels of IL-1β, IL-6 and TNF-α and decreased IL-4 expression were found in HG-induced HK-2 cells, and this effect could be suppressed by the transfection of miR-218 inhibitor. However, the transfection of si-SPRED2 or the treatment of 3-MA reversed the effect of miR-218 inhibitor on inflammatory factor secretion in HG-induced HK-2 cells (Figure 2D,E). The results of ANOVA test are listed in Table 2. All the above results suggested that the inhibition of SPRED2 or autophagy reversed the effect of miR-218 inhibitor on inflammatory factor secretion and oxidative stress in HG-induced HK-2 cells.
Inhibition of SPRED2 or autophagy reverses the suppressive effect induced by miR-218 knockdown on cell apoptosis in HG-induced HK-2 cells
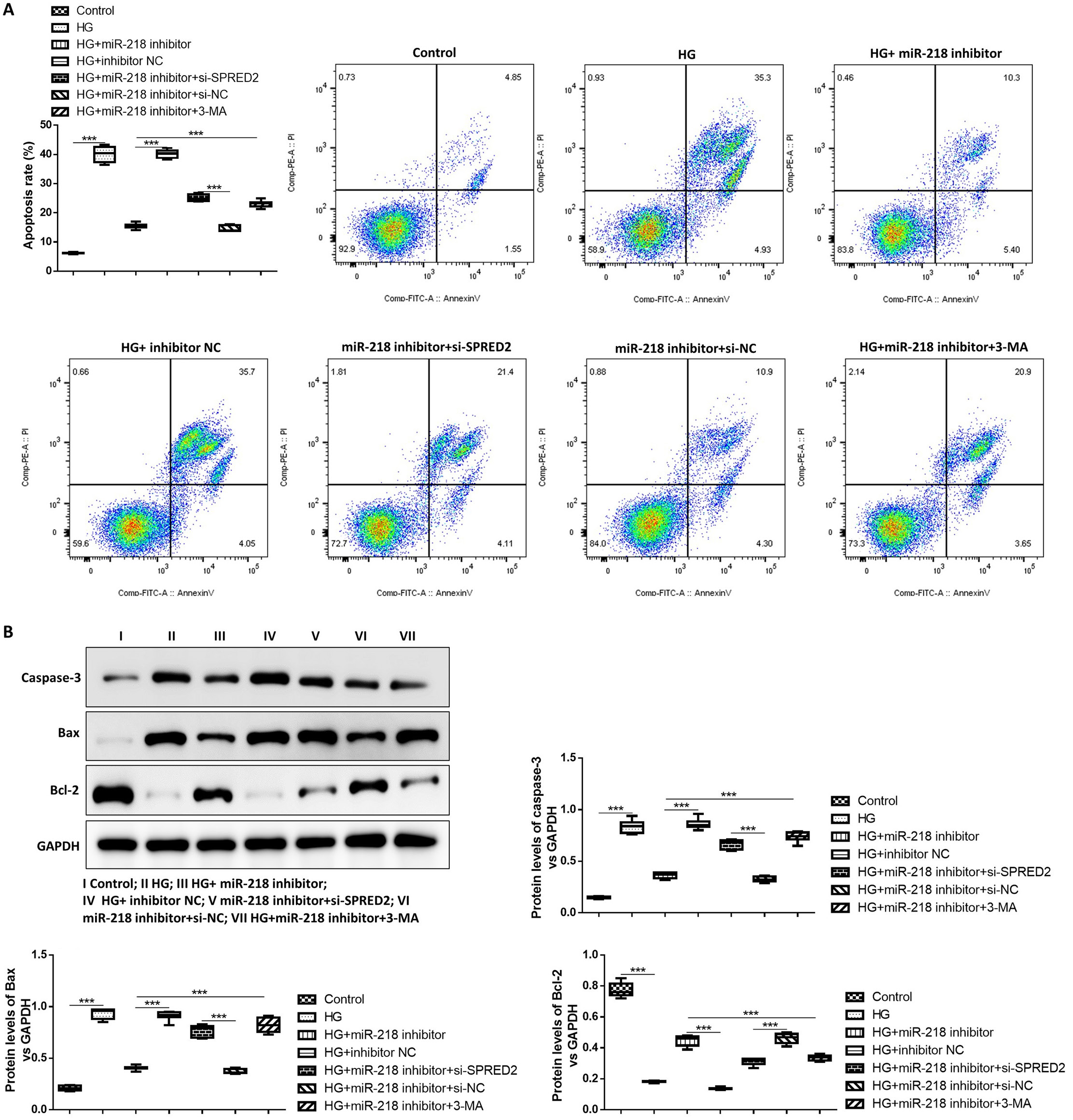
The effect of miR-218 and SPRED2 on apoptosis in HG-induced HK-2 cells was also studied. We observed that miR-218 knockdown attenuated HG-induced cell apoptosis, which could be reversed by SPRED2 silencing or 3-MA treatment (Figure 3A). The expression of apoptosis-related protein was detected to confirm this result. The level of cleaved caspase-3 and Bax was significantly elevated, and Bcl-2 expression was greatly decreased in HG-induced HK-2 cells. The miR-218 knockdown obviously suppressed the level of cleaved caspase-3 and Bax, but increased Bcl-2 expression in HG-induced HK-2 cells; however, this effect was alleviated by silencing SPRED2 or 3-MA treatment (Figure 3B). The results of ANOVA test are listed in Table 3. These findings proved that the inhibition of SPRED2 or autophagy reversed the inhibitory effect of miR-218 silencing on cell apoptosis in HG-induced HK-2 cells.
Inhibition of SPRED2 or autophagy reverses the effect induced by miR-218 knockdown on autophagy in HG-induced HK-2 cells
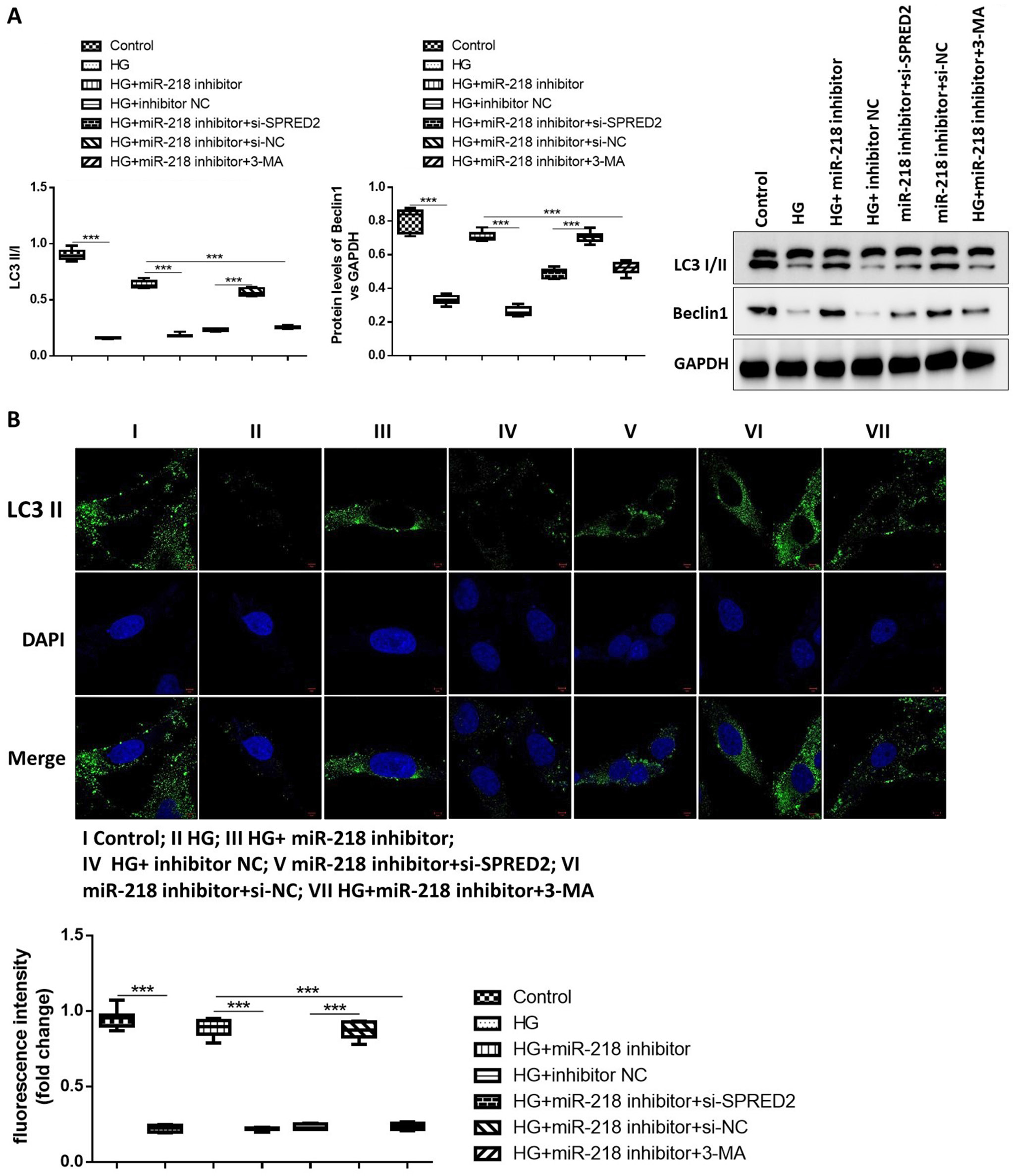
Finally, the role of miR-218 and SPRED2 on autophagy in HG-induced HK-2 cells was studied. The effect of si-SPRED2#1 and si-SPRED2#2 on cell autophagy was analyzed and the result suggested that the transfection of both siRNAs inhibited cell autophagy and si-SPRED2#2 showed more significant effects (Supplementary Figure 1). Autophagy-related protein level of Beclin 1 and the conversion rate of LC3-II/I were detected with western blotting (Figure 4A). The HG induction obviously suppressed Beclin 1 expression and conversion ratio of LC3-II/I, which was increased by miR-218 inhibitor. However, si-SPRED2 or 3-MA alleviated the effect of miR-218 inhibitor on autophagy-related protein expression. Immunofluorescence result showed that the LC3-II expression was downregulated by HG induction, but it was upregulated by miR-218 knockdown. Moreover, downregulated SPRED2 or 3-MA reversed the regulation of miR-218 on LC3-II expression (Figure 4B). The results of ANOVA test are listed in Table 4. All these findings indicated that autophagy activated by silencing miR-218 was suppressed by SPRED2 knockdown or autophagy inhibitor.
Discussion
Around 20–40% of DM patients develop DN, and the 5-year survival rate of patients with end-stage renal disease (ESRD) is as low as 20%.26, 27, 28, 29 Recently, autophagy and oxidative stress have gradually become recognized as a new pathogenesis of DN and draw more and more attention.30, 31, 32 In this study, we investigate the effects of miR-218/SPRED2 axis on oxidative stress and inflammatory response in HG-induced renal tubular epithelial cells.
In the last decade, numerous evidence surfaced for variety of miRNAs involvement in DN development. A clinical study found that miR-29c regulated the expression of inflammatory cytokines in DN by targeting tristetraprolin.33 The role of miR-218 in DN was also illustrated in several in vivo and in vitro studies.34 An in vitro study showed that downregulated miR-218 suppressed the level of inflammatory factors and attenuated the HG-induced injury in renal proximal tubule cell.35 Moreover, miR-218 expression was upregulated in HG-treated podocytes and miR-218 silencing inhibited apoptosis in HG-treated podocytes.36 Upregulated miR-218 increased the expression of pro-inflammatory cytokines, decreased the expression of anti-inflammatory cytokines and accelerated the process of epithelial–mesenchymal transition in HG models.37 All of these studies indicate that miR-218 facilitates inflammation response and oxidative stress in DN, and thus promotes DN development. The present study also found that miR-218 expression was upregulated in HG-induced HK-2 cells, and that downregulated miR-218 alleviated cell apoptosis, inflammation and oxidative stress, and enhanced autophagy in HG-induced HK-2 cells. However, a DN rat model revealed that overexpressing miR-218 was sufficient to alleviate renal injury.38 This issue needs deep insight in further study.
Autophagy is considered a stress-responsive intracellular system that plays an essential role in promoting cell against hypoxia, endoplasmic reticulum stress or oxidative stress, associated with the pathogenesis of diabetes-related diseases.39, 40, 41, 42 In DN development, cell autophagy also plays important roles, affecting cell apoptosis and inflammation of renal tubular epithelial cells.43, 44 Generally, the dysfunction of autophagy is considered a contributor to DN and renal injury.45, 46 Kitada et al. demonstrated that autophagy activation might be a potential therapeutic option for DN.47 Meanwhile, a previous study discovered that impaired autophagy in podocyte cells could accelerate renal damage.48 The SPRED2 was identified as an activator of autophagy and SPRED2 deficiency might affect autophagy, resulting in cardiac dysfunction and life-threatening arrhythmias.49 Interacting with LC3, SPRED2 promoted autophagosome maturation, thereby leading to cell death in tumor.50 Based on bioinformatics analysis, SPRED2 was predicted to be one of the target genes of miR-218 in our study. However, no studies illustrate the regulation of expression between miR-218 and SPRED2 in renal tubular epithelial cells; besides, the molecular mechanism action of SPRED2 in DN was not reported. In a recent study, miR-218 was found to regulate SPRED2 expression in PC12 cells.51 Therefore, we hypothesized that miR-218 might promote the development of DN by inhibiting SPRED2-mediated autophagy. Our results showed that miR-218 negatively regulated SPRED2 expression in HK-2 cells. We demonstrated for the first time that SPRED2 suppression reversed the effect of downregulated miR-218 on inflammatory factor secretion and oxidative stress, apoptosis and autophagy.
Limitations
The main limitation of this study is that it lacks in vivo evidence. The signaling path ways regulating miR-218 in DN are also unclear.
Conclusions
In summary, miR-218 was upregulated and SPRED2 was downregulated in HG-induced renal tubular epithelial cells. The miR-218 negatively regulates SPRED2 expression, but SPRED2 knockdown reverses the effect of downregulated miR-218 on inflammatory factor secretion and oxidative stress, apoptosis and autophagy. We demonstrated for the first time that miR-218 promoted oxidative stress and inflammatory response in HG-induced renal tubular epithelial cells by inhibiting SPRED2.
Data availability
The Supplementary data are available at https://doi.org/10.5281/zenodo.6383061. They consist of 3 files:
1. Statistical Data is the original file from the SPSS software;
2. Tables of Statistical Data contain the statistical results for calculation of all data in the figures;
3. Supplementary Figure shows the effects of transfection of si-SPRED2#1 and si-SPRED2#2 on cell autophagy.