Abstract
Heart failure (HF) is a pathophysiologically complex disease that is exceptionally heterogeneous in terms of its etiology. It is associated with unsatisfactorily high mortality, both in-hospital and post-discharge, as well as with very frequent rehospitalizations. High phenotypic variability, coexistence of various hemodynamic disorders (such as changes in systemic and pulmonary vascular resistance, increased central venous pressure, impaired heart cardiac output, and fluid overload) and coexisting metabolic and neurohormonal disorders may eventually lead to impaired systemic perfusion. Congestion that impairs renal perfusion has a significant impact on both glomerular filtration and the renal tubular function. This review article discusses the importance of changes caused by HF in various nephron segments, phenotyping of cardiorenal syndromes, the role of effective natriuresis in decongestion, and the importance of known and new diagnostic biomarkers in predicting renal dysfunction. A better understanding of cardiac and renal interactions may help in selecting an effective, efficient and nephroprotective strategy of treatment for patients with HF.
Key words: heart failure, cardiorenal syndrome, worsening renal function, renal markers, natriuresis
Introduction
Mutual hemodynamic and neurohormonal interaction of the heart and kidneys determines the exceptional dependence of both organs. Heart failure (HF) and kidney disease (KD) often coexist, which carries significant prognostic implications. Chronic kidney disease (CKD), defined as lowering of estimated glomerular filtration rate (eGFR) <60 mL/min/1.73 m2 is present in 4.5% of general population and as many as 50% of patients with HF.1, 2 A large meta-analysis involving over 1,000,000 HF patients revealed that the presence of CKD doubles the risk of general mortality.2
Objectives
Modifications of diuretic treatment in patients with acute HF (AHF), associated with the use of both too high and too low doses of loop diuretics, result in excessive dehydration, risk of hypotension and worsening renal function. The aim of this review article was to evaluate and compare information about the important role of renal tubular function in the context of natriuresis and chloride metabolism associated with effective diuresis, including a comprehensive assessment of cardiorenal syndrome (CRS) and renal biomarkers. An optimal diuretic treatment, comprising different diuretic classes and their various activities, should be applied in patients with AHF.
Materials and methods
This review article provides an overview of current publications indexed in MEDLINE database (using PubMed and EBSCO), Scopus and Embase until July 2021. Taking into consideration the selected studies, the following keywords and Medical Subject Headings (MeSH) were used: “heart failure”, “natriuresis”, “spot urine sodium”, “cardiorenal syndrome”, “worsening renal function”, and “acute kidney injury”. The Boolean operators “AND” and “OR” were also used to narrow the search. The term “heart failure and worsening renal function” appeared in 1966 (Scopus), 1403 (PubMed) and 1223 (Embase) results, respectively. Additionally, a detailed search for articles was carried out using the keywords “heart failure AND cardiorenal syndrome AND acute kidney injury”, which provided 297 results (51 in 2020 and 2021) in PubMed, 264 results (Scopus) and 328 results (Embase), respectively. Using the keywords “heart failure AND natriuresis” resulted in 1054 (Scopus), 969 (PubMed) and 1408 (Embase) articles found, respectively. In PubMed, the term “heart failure AND spot urine sodium” resulted in 33 articles (out of which 9 were published in 2020 and 2021), 50 articles in Scopus and 17 results in Embase. Due to the large number of articles related to the term “renal markers AND acute kidney injury”, we had to narrow the search to selected renal biomarkers. In order to achieve a comprehensive pathophysiological approach, in addition to using selected keywords, we also performed a manual search.
Renal perfusion compared to oxygen use: differentiated distribution of energy expenditure
The kidneys are the most vascularized organ of the body. Renal blood flow (RBF) constitutes 20–25% of resting cardiac output (CO), and involves the renal cortex and medulla whose dynamics and metabolism differ.3 In terms of anatomy, renal corpuscles (glomeruli surrounded by Bowman’s capsule) and their vascularization are inside the cortex, while renal tubules, located outside the cortex, reach the medulla. Physiologically, the kidneys show a high demand for oxygen (O2), but it is very different in the cortex and medulla. Perfusion of the cortex constitutes as much as 90% of blood flow and is autoregulated at low and normal oxygen consumption (VO2), which is first and foremost associated with the use of O2 in renal tubules, necessary for the reabsorption of approx. 99.5% of filtered sodium. At the level of the medulla, blood flow is low and well-maintained even when systemic perfusion is significantly reduced.4, 5 In most tissues (e.g., in the brain), increased O2 demand is followed by increased perfusion and O2 supply. However, the load of reabsorbed sodium increases in the kidneys simultaneously with increased RBF and increased O2 supply, which causes further increase in O2 consumption and results in a high energy expenditure of the kidney. As a consequence, all of these processes may paradoxically cause hypoxia of intrarenal tissue and renal tubules (such a mechanism may occur in the case of hypertensive and diabetic nephropathy).5 On the other hand, decreased CO, lowering of RBF in AHF and activation of the compensatory mechanisms (sympathetic nervous system (SNS) and renin–angiotensin–aldosterone system (RAAS)) result in poor renal perfusion, hypoxia of cells and renal tubules, as well as further damage thereto.4
The role of individual
nephron segments in water
and electrolyte balance
From the clinical perspective, renal metabolic function of the kidneys understood in the context of nephron activity should be analyzed in 3 anatomical and functional areas: glomeruli (filtration function), renal tubules (tubular function associated with reabsorption of tubular fluid with electrolytes with simultaneous secretion) and renal interstitium (absorption of water and electrolytes into peritubular capillaries).
Filtration function
The eGFR is the total result of the number of functional nephrons. Under physiological conditions, the pace of glomerular filtration of a single nephron is 40–70 nL/min with filtration fraction (FF) of 20–25%. With normal renal perfusion, hydrostatic pressure in glomerular capillaries slowly increases from the proximal to the distal end. As a result, the gradient of ultrafiltration pressure is equally maintained along the entire glomerular capillary.5, 6, 7 The total volume of primary urine produced under normal conditions is approx. 180 L per day, containing approx. 1.5 L of NaCl, most of which is reabsorbed into renal interstitial fluid and systemic circulation. The primary filtrate contains electrolytes (sodium, chloride) and nitrogen metabolism products, urea, creatinine, uric acid, as well as mineral salts and amino acids.1, 8 Less than 1% of NaCl and only a small part of other dissolved substances are excreted with final urine. It is worth noting that glomerular filtration reflects only the filtration function of the nephron, which is very important in the production of primary urine. The formation of final urine and effective diuresis, understood as aquaresis and natriuresis as well as normal regulation of chloride concentration, stem from the efficiency of renal tubule function.1, 6
Tubular function
Proximal tubule
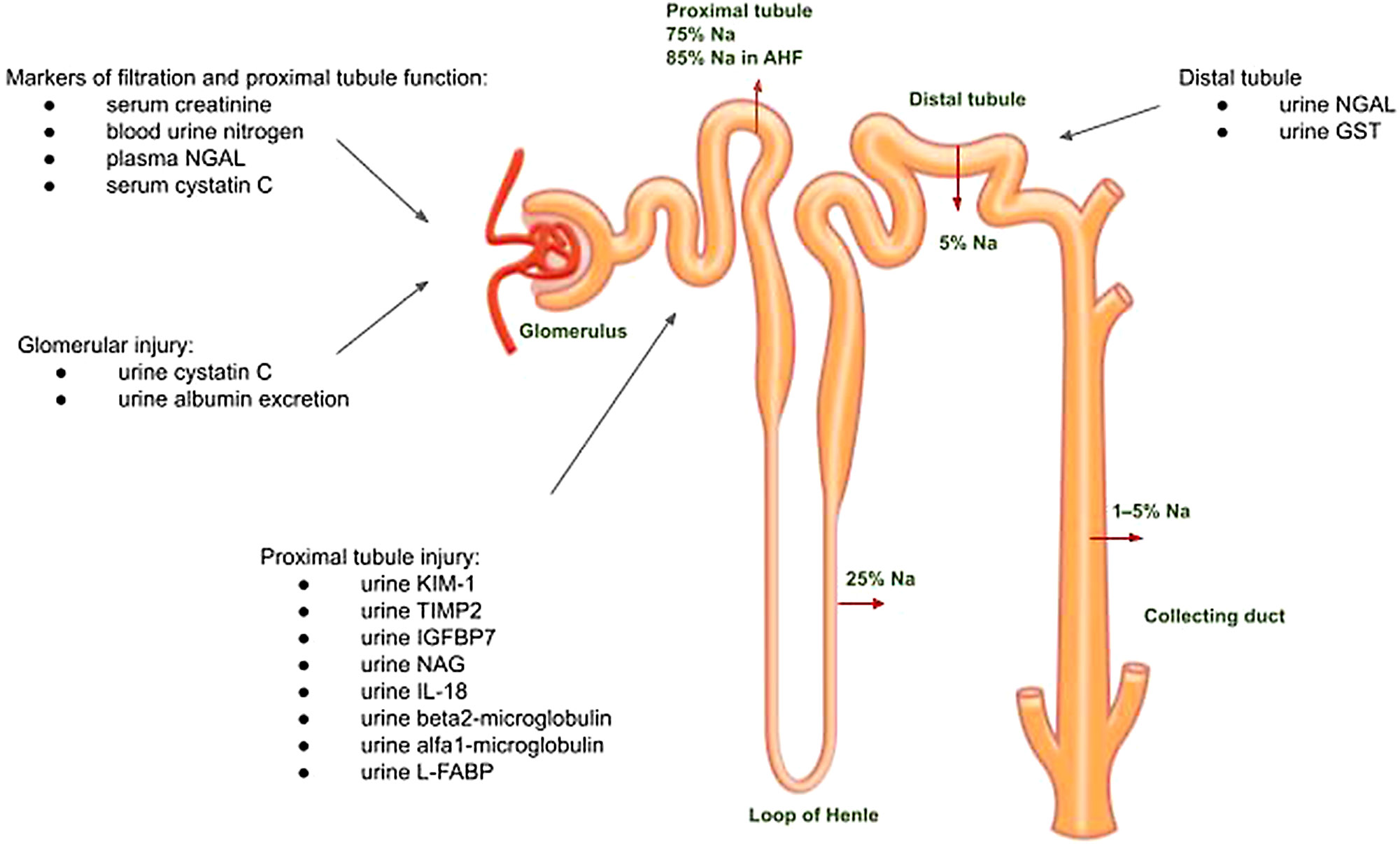
Under normal conditions, the wall of the proximal tubule is freely permeable to water, which is accompanied by the reabsorption of sodium to maintain osmotic balance. Various transporters in the wall of the proximal tubule are responsible for such active transport (e.g., Na+/K+-ATPase of the basolateral membrane). This relatively stable sodium fraction (~75%) is reabsorbed in the proximal segment of the nephron and determined by Starling forces, regardless of neurohormonal activation. As a result of functional changes in the proximal renal tubule, as much as 85% of sodium can be reabsorbed in AHF (Figure 1).8
Loop of Henle and distal tubule
Roughly 25% of sodium is then reabsorbed again in the site of action of the Na/K/Cl cotransporter in the thick ascending loop of Henle’s limb. It is the site of action of loop diuretics (blocking of the cotransporter leads to an increase in the sodium load in the distal tubule and promotion of the excretion of sodium, chloride and – to a lesser extent – potassium). In the distal convoluted tubule, the sodium-chloride cotransporter is responsible for only about 5% of sodium reabsorption. Since this is the site of action of thiazide diuretics, the diuretic effect of these medications is poorer than that of loop diuretics. In the collecting duct, an aldosterone-dependent cotransporter is active (and responsible for the absorption of 1–5% of sodium), which is inhibited by mineralocorticoid receptor antagonists.6
Chloride and tubuloglomerular feedback
Chloride is freely filtered by glomeruli. Approximately 60% of chloride is absorbed by the proximal renal tubule. Its concentration in urine depends proportionally on sodium concentration and is regulated by a number of transport processes.9 Macula densa (MD), which is responsible for physiological tubuloglomerular feedback in the kidney and plays a role in sodium and chloride concentration regulation, is important for efficient function of the organ. It is located in the place where the distal tubule approaches the renal corpuscle and acts as an osmoreceptor of urine flowing through the distal convoluted tubule.6 The MD reacts to changes in chloride concentration and the signal indicating changes in osmotic pressure are transmitted to the afferent glomerular arteriole. Describing the so-called “chloride theory”, Kataoka draws attention to the essential role of chloride in the pathogenesis of congestion in AHF. Low chloride load (along with low sodium load) in the urine of patients with AHF, in whom there is an increased sodium resorption in the proximal tubule, stimulates the excretion of renin by juxtaglomerular cells. Renin causes the dilation of the glomerular afferent arteriole, the hyperfiltration of glomeruli and, as a result, the activation of the vicious circle of the RAA.10, 11 In turn, an increase in the influx of sodium and chloride into the distal tubule activates tubuloglomerular feedback by the breakdown of adenosine triphosphate (ATP) into adenosine, which leads to the constriction of the afferent glomerular arteriole and stops the activation of the RAA. Thanks to tubuloglomerular feedback, the nephron is also protected against hyperfiltration.6, 12
Renal interstitium
In AHF, increased FF, which can be present even before the occurrence of a significant decrease in eGFR, causes significant changes in both hydrostatic and colloidal osmotic pressure in peritubular capillaries as well as in the interstitial portion of the kidney. Due to the fact that the kidneys are surrounded by a fibrous and fatty capsule and the renal fascia, hydrostatic pressure is evenly increased in the lumen of peritubular capillaries and renal interstitium (renal venous hypertension), while the oncotic pressure of the interstitial fluid decreases due to increased flow of lymph, which flushes out interstitial proteins. On the other hand, peritubular capillaries are impermeable to plasma proteins, which explains why intracapillary colloidal osmotic pressure remains high. These mechanisms facilitate passive resorption of sodium with resorption of water.6, 13
Natriuresis and diuretic
treatment strategy
Early measurement spot urine sodium (UNa) may be useful in identifying the patients who show poor response to diuretic treatment. In a prospective observational study, UNa was measured after the administration of the first dose of an intravenous diuretic medication. The risk of composite primary endpoint (death from any cause after 90 days, the use of a mechanical circulatory support system at hospital admission and inotropic agents at discharge) was twice as high in patients with UNa ≤ 60 mmol/L. Worsening renal function (WRF) occurred significantly more often in patients with UNa < 60 mmol/L.1, 14 In a Renal Optimization Strategies Evaluation (ROSE)-AHF study, a lower value of UNa ≤ 60 mmol/L within the first 24 h of diuresis was characteristic of patients in whom there was a risk of extended hospitalization.15 In addition, the evaluation of renal function, understood as eGFR, does not provide reliable information on sodium balance.16 Maintained eGFR profile (60 mL/min/1.73 m2) in patients with HF and UNa ≤ 60 mmol/L is associated with high annual mortality rate.16, 17 Early monitoring, consisting in the recording of UNa 50–70 mmol/L after 2 h and/or diuresis 100–150 mL within the first 6 h of treatment, usually makes it possible to identify patients showing an inadequate response to diuretic therapy.1, 18, 19 Therefore, it is necessary to interpret changes in renal function adequately early to identify patients in whom there is a risk of WRF and a lack of clinical improvement.20, 21 Furthermore, in AHF, sodium excretion is related to clinical status and has a different
prognostic value between early phase of hospitalization and at discharge.22 As a result, the physician will be able to make the decision of modifying the dose of the loop diuretic (double the dose) in order to obtain maximum plasma concentrations of the medication, or using sequential nephron blockade, i.e., combining the treatment with diuretics with a different mechanism of action.1
Phenotypes of CRS
In some HF patients, acute kidney injury (AKI) leads not only to transient but also permanent impairment of glomerular filtration. Apart from hemodynamic mechanisms (impaired organ perfusion due to decreased CO and high central venous pressure (CVP)), an important role is played by the dysfunction of SNS, activation of the RAAS, disruption of the hypothalamic–pituitary axis, and stimulation of inflammatory reactions, including changes in cytokine signalling.23, 24 It should be noted, however, that AKI may be iatrogenic and associated with suboptimal dosing of diuretics in patients with HF. Such treatment may increase the risk of excessive decongestion by decreasing intravascular volume, with secondary development of prerenal failure. From the clinical perspective, it is very important to identify this phenomenon quickly, for example by using early biomarkers of renal tubular injury.25 This strategy may play a key role in the appropriate modification of treatment and prevention of the development of CRS.
CRS type I
Sudden worsening of the CO (caused by myocardial ischemia, arrhythmia, severe valvular heart diseases, or myocarditis), that results in a cascade of hemodynamic and neurohormonal changes, leads to the arterial underfilling.26 It causes RBF reduction, secondary activation of the compensatory mechanisms of RAAS and an increase in SNS tension. Angiotensin II activity results in the constriction of the efferent glomerular arteriole. In order to maintain optimal renal perfusion and constant FF, through an increase in the expression of vasodilators (prostaglandin, nitric oxide), the afferent glomerular arteriole is dilated.6, 26, 27 An increase in FF initially masks the absolute decrease in eGFR, but after reaching the maximum FF at the value of ~60%, further RBF decrease causes a significant linear decrease in eGFR. Ultrafiltration pressure gradient cannot be maintained any longer at the entire length of the glomerular capillary, which partially loses its ultrafiltration efficiency (the “wasted capillary” phenomenon occurs). It leads to a decrease in the general number of functionally active nephrons, causing a decrease in glomerular filtration.1 Reactively increased secretion of vasopressin results in reverse resorption of water from the collecting duct of the nephron.3 Increased reflex adrenaline secretion causes a significant increase in renal vascular resistance, which leads to further impairment of renal perfusion. In addition, the expression of endothelin I (which is a substance with strong vasoconstrictive properties) leads to vasoconstriction, fostering remodeling processes and renal vascular fibrosis.28 Also, abnormalities in the venous area of renal capillaries, stemming from increased CVP, lead to renal interstitial edema, hypoxia and dysfunction of renal tubules (resulting from increased hydrostatic pressure and impaired outflow of blood from the kidneys). In a similar mechanism, increased intra-abdominal pressure (IAP), occurring, for example, in the case of coexisting ascites, leads to an impaired renal perfusion due to compression. This is due to a decrease in abdominal perfusion pressure, which is the difference between mean arterial pressure and IAP.29
CRS type 2
Type 2 CRS is developed secondary to chronic HF, in the case of which we observe gradual, progressive activation of compensatory mechanisms (stimulation of RAAS, SNS and endothelin overproduction). These processes lead to worsening of renal perfusion through the development and perpetuation of structural changes in the renal parenchyma, renal vascular fibrosis as well as dysfunction of renal tubules. Apart from its vasoconstrictive effect, endothelin 1 production contributes to low urine sodium excretion, the occurrence of inflammation in the kidneys and increased expression of aldosterone. Moreover, the coexisting synthesis of fibronectin stimulates fibrotic processes that are frequently irreversible.30 Aldosterone shows expression in preglomerular vessels, glomeruli and along distal tubules. Its increased production leads to vascular remodeling, glomerular sclerosis, generalized endothelial dysfunction, and increased oxidative stress.31 The CRS has been divided into 5 subtypes (Figure 2).3
CRS: the updated concept
The AHF is a complex syndrome involving numerous organs, including kidneys.32 The kidney is an important homeostatic organ and its activity is closely interrelated with the function of the cardiovascular system.24 Although CRS subtypes systematize the knowledge that allows for research on the interaction between the cardiovascular system and the kidneys, it is only a simplification that ignores the interaction between the kidneys and other key organs and systems (lungs, liver, skeleton, gastrointestinal tract and its microbiome, SNS, adipose tissue, skeletal muscles).33, 34 It is crucial to notice the role of the kidneys in the metabolism of carbohydrates35 as well as in the regulation of oxygenation of all tissues through their participation in the metabolism of iron and hemoglobin.36 This homeostatically central role of the kidneys means that a dysfunction in any of these organs may have an impact on kidney function and such impact may be responsible for the differentiation of the renal response to various cardiovascular diseases.
Since the definition of CRS was developed, the understanding of the heterogeneity of HF, especially the role of other diseases and organs beyond the circulatory system, has increased significantly.37 For this reason, the analysis of renal function within the conceptual paradigm of the CRS has exhausted all its options. There is a need for an interdisciplinary concept in which it will be possible, on the one hand, to understand the causes of high heterogeneity of renal function in HF and during its treatment, and on the other hand, to identify such functional renal responses and the circumstances of their emergence that are clinically unfavorable. Regardless of the phenotype of HF, the functional renal response to its presence and treatment may fall into one or a combination of the following clinically important categories:
− inadequate control of the extracellular fluid volume with different responses to the diuretics,
− decrease in glomerular filtration,
− decrease in diuresis,
− hyperkalemia/hypokalemia,
− hyponatremia,
− acidosis/alkalosis with hypochloremia.
The abovementioned dysregulation involving the kidneys occurs in everyday medical practice and it is difficult to relate it to the established concepts of KD, which includes its 2 types (acc. to KDIGO 2012):
1. CKD with eGFR below 60 mL/min/1.73 m2 for 3 months or eGFR over 60 mL/min/m2 with additional evidence of kidney injury:
− albuminuria/proteinuria,
− macroscopic or microscopic changes,
− etiology (diabetes, hypertensive, interstitial disease, etc.).
2. AKI with an increase in serum creatinine concentration (SCr) by ≥0.3 mg/dL (26.5 µmol/L) within 48 h, or a ≥1.5-fold increase within the previous 7 days, or a decrease in diuresis by <0.5 mL/kg/h within 6 h.
These 2 types of KD can overlap, and this is very common in a significant percentage of patients with HF. However, these syndromes do not include abnormalities in fluid and electrolyte balance, fluid overload and resistance to diuretics. In clinical practice, we see patients with HF with preserved or reduced ejection fraction with concurrently impaired glomerular filtration, hyperkalemia or hypochloremic alkalosis due to the use of diuretics.
WRF in the course of HF
The AKI is associated with an increased risk of hospitalization and high in-hospital and post-discharge mortality.38, 39 It is important to note that the AKI is a clinical syndrome and is not synonymous to acute renal failure (ARF). The coexistence of a rapid, persistent worsening renal function, increased concentration of renal injury markers, accumulation of nitrogenous and non-nitrogenous metabolic products, as well as profound electrolyte disturbances leads to ARF. Based on, for example, the Risk, Injury, Failure, Loss, End-stage renal disease (RIFLE) criteria, we can calculate risk and damage and predict gradual progression of renal disease to end-stage renal failure. However, they only make it possible to characterize successive stages of renal failure development, without necessarily showing sensitivity in the detection of early intrarenal dysfunction.3 Unfortunately, there are patients who remain at risk of developing progressive CKD, despite complete or nearly complete restoration of renal function after an AKI episode. The occurrence of AKI in patients previously diagnosed with CKD is associated with accelerated progression thereof as well as the occurrence of end-stage renal failure. The AKI occurs mainly in HF patients hospitalized due to AHF. The incidence of AKI in population is ~20%.40, 41, 42 The significance of WRF among patients with AHF has been emphasized for years.40, 43 The presence of WRF, understood as an increase in SCr > 0.3 mg/dL in serum or a decrease in eGFR by >25% (expressed in mL/min/1.73 m2, calculated according to the Modification of Diet in Renal Disease (MDRD) formula), together with worsening or lack of improvement in signs and symptoms of AHF, is known as the “true WRF”. This condition requires intensification and optimization of pharmacological treatment (and, in some cases, the introduction of renal replacement therapy), especially when resistant fluid overload and metabolic disorders (hyperkalemia and acidosis) are also present, and is associated with worse prognosis. Authors of one study observed that general mortality increased in a 12-month follow-up of patients with true WRF (in a population of 266 patients with HF, 73 patients (27%) reached endpoint). In the group of patients with true WRF death occurred in 9 out of 11 (82%) patients, in the group with pseudo-WRF in 3 out of 27 (11%) patients and in the group without WRF in 61 out of 228 patients (27%) (p < 0.001).20, 21 Importantly, in patients with simultaneously preserved diuresis, effective decongestion and improvement in clinical condition during the course of treatment, an increase in creatinine/decrease in eGFR does not imply worse prognosis (“pseudo WRF”).1, 21 It is also worth noting that the administration of loop diuretics, angiotensin-converting enzyme (ACE) inhibitors or aldosterone antagonists before hospitalization due to HF does not have an impact on an increased risk of WRF. Pseudo-WRF may occur as a result of the implementation of a targeted therapy reflecting the neurohormonal blockade, understood as the inhibition of the RAA system, and it is not necessarily a sign of direct renal dysfunction. Only isolated changes in SCr occur in this group of patients.1, 2 Therefore, a consistent evaluation of renal function must be conducted in each patient suffering from HF, especially from AHF. Such strategy makes it possible to assess the effectiveness, efficacy and – thus – safety of the treatment. Therefore, researchers are still seeking markers that are sensitive, easy to determine and available that will be helpful in identifying patients with sub-clinical AKI who have an increased risk of adverse outcomes. There are some systems that prevent intravascular hypovolemia during decongestion, which may reduce the risk of WRF; however, this issue needs further investigation.44 The safety of ultrafiltration also needs to be clarified in the future.45
Biomarkers of AKI
Monitoring of SCr, as a common endogenous marker of glomerular filtration function, is a standard in the evaluation of renal function. This method, however, has a number of limitations. Creatinine, a product of creatine metabolism, which is a reserve of high-energy phosphates in skeletal muscles, is freely filtered by renal glomeruli, without being resorbed or secreted in the kidney (except for 15% creatinine in urine which comes from tubular secretion in the proximal tubule through excretion of organic cations).4 When eGFR is reduced, its half-life extends from 4 h to even 24–72 h. It is particularly important in the context of impaired metabolism in patients with end-stage renal disease (ESRD), in whom creatinine can be excreted or metabolized via extrarenal pathways (up to 66% of daily production), most likely by intestinal flora.46, 47 Therefore, SCr may increase only after 24–36 h despite early renal injury, which significantly delays the diagnosis of AKI.1, 7, 47, 48 There are also reports of delayed increases in SCr despite changes in eGFR occurring already at an early stage of AKI, which can only be visible after a 50% loss of renal functional capacity.49 In addition, SCr depends not only on glomerular filtration function, but also on factors such as sex, age and body weight. In patients with low muscle mass, features of cachexia, liver diseases or sepsis, SCr is lower and, as a result, eGFR may be overestimated. A protein-rich diet – consuming large amounts of boiled meat – leads to an increase in SCr caused by increased intestinal resorption of creatinine from consumed meat.1, 7, 46, 47, 48 Thus, an increase in its SCr is not specific for early renal tubule damage (but is rather an effect of the loss of glomerular filtration function) and requires consideration of prerenal and extrarenal causes.47, 49 Blood urea nitrogen (BUN) evaluation is also worth mentioning, which is also helpful in diagnostic evaluation of moderate and severe CKD, regardless of eGFR. It has also been shown that increased levels of BUN in AHF patients, even those in whom SCr levels are normal or slightly elevated, are correlated with increased mortality. The necessity to take into account SCr and the inertia of analysis resulting thereof, constitutes a limitation of the method.50 General urinalysis is a widely available clinical parameter with diagnostic value in the evaluation of structural and functional causes of KD. The evaluation of urine sediment examination, based on the evaluation of active sediment, the number and type of epithelial cells or casts, is significant in diagnosis of KD. This interpretation, however, frequently does not indicate precisely whether the disease is acute or chronic.3 Renal function should also be considered when evaluating the clinical significance of urine osmolality. Low urine osmolality is an independent risk factor for CKD progression, and its predictive ability is not superior to eGFR.51
Novel renal biomarkers
In the last decade, a very interesting diagnostic platform appeared among urinary markers that are of significance in early prediction of AKI. Such biomarkers can be divided into several groups according to their biological origin and time of release after renal injury (Table 1), or potentially reversible or irreversible effects on AKI (Figure 3).52
Neutrophil gelatinase-associated lipocalin
The best known marker is the neutrophil gelatinase-associated lipocalin (NGAL), which is a sensitive and specific urine and serum biomarker for predicting AKI at an early stage. A 10-fold increase in NGAL serum concentration and over a 100-fold increase in its concentration in the urine of adults has been observed in AKI.49, 53 In a meta-analysis of 10 studies involving 2000 patients with CRS, an increase in serum and urine level of NGAL were predictive for renal replacement therapy and death.3 What is more, an increase in serum NGAL indicates AKI progression after cardiac surgeries, secondary to post-contrast AKI, as well as septic shock in children.49 Importantly, its high level is observed in the case of inflammation, as NGAL is an acute phase reactant and can be released from neutrophils and macrophages.54 Therefore, in the context of diagnostic evaluation and intervention in early renal tubular injury, the dynamics of NGAL synthesis, both in the serum and urine, can be a useful tool for a noninvasive evaluation of AKI, even before the development of clinically evident form of the disease.53
Kidney injury molecule-1
In ischemic or nephrotoxic kidney injury, kidney injury molecule-1 (KIM-1) is produced and accumulated in very large amounts on the membrane of the renal proximal tubules. Studies have shown an increase in KIM-1 expression in kidney biopsy samples with confirmed acute tubular necrosis. An increase in the concentration of this biomarker in urine is crucial, especially in ischemic AKI, compared to other causes (e.g., in the case of post-contrast nephropathy or CKD, even despite concomitant urinary tract infection).55 Studies also suggest that the urinary KIM-1 may constitute a predictive factor for the introduction of renal replacement therapy and causes a risk of death during hospitalization in patients with AKI. However, studies available to date are not sufficient to determine the cutoff value that is predictive for AKI in the setting of intensive medical care.56
Cystatin C
In a study involving 444 patients in an intensive care unit (ICU), cystatin C (Cys C) concentration in urine was significantly higher in patients with sepsis or AKI. However, results of studies investigating the use of Cys C serum concentration in the detection of AKI in patients in ICU were conflicting.54, 57, 58 Therefore, Cys C (primarily due to its constant rate of secretion in the organism) is a good, early marker of GFR (without the impact of infection or liver diseases). Serum Cys C concentration also does not depend on age, sex, race, muscle mass, and hydration. It is not, however, specific in diagnostic evaluation of AKI.54
Other biomarkers that are worth mentioning include tissue inhibitor of metalloproteinases (TIMP-2) and insulin-like growth factor-binding protein 7 (IGFBP7). They are available for clinical use in the USA. In 728 critically ill patients not showing any signs of AKI, the determination of urinary levels TIMP-2 and IGFBP7 constituted a predictive value for the development of AKI (p < 0.002).3 The abovementioned renal markers in AKI have been summarized in Figure 2.
Features of a perfect renal biomarker
Due to the limitations discussed above, researchers are constantly seeking an ideal diagnostic biomarker for predicting AKI. It should be characterized by, among other things, high specificity for the kidneys, the ability to distinguish types, duration and etiology of AKI (renal injury, including tubular and glomerular, prerenal and extrarenal AKI). It is recommended that its increase provide early information about kidney dysfunction and illustrate the scale of such damage (with an established threshold for the assessment of dysfunction progression and regression). It should also be widely available in everyday clinical practice and be easy, precise and quick in terms of determination, as well as inexpensive.49 Despite intensive search, an ideal marker for early diagnosis of AKI has not been found yet, which seems understandable due to structural heterogeneity of the kidneys.
Classification of diuretics:
clinical perspectives
Using the selected renal biomarkers may be helpful in predicting AKI, but there is no evidence on their effect on diuresis. The novel CRS-guided approach, which takes into account the importance of metabolism of both sodium and chloride, is important to regulate plasma volume, diuresis and natriuresis during decongestive treatment in AHF. The pathophysiological mechanisms in various nephron segments explain the crucial role of these electrolytes and highlight the need for proper choice of a combination of the different diuretic classes. Dyschloremia is postulated as one of the main causes of worsening HF during decongestive treatment.59 A new classification of diuretics based on their effect on serum chloride concentration has been proposed in recent years. This concept is consistent with recent clinical observations that chloride-regaining diuretics preserve plasma volume, enhance vascular “tonicity” and avoid diuretic resistance (Table 2).60
Study limitations
The study lists selected renal biomarkers which are gaining increasing interest in research, but listing them all would exceed the scope of this publication. In addition, the review was limited to selected pathophysiological mechanisms related to the interdisciplinary complex issue involving cardionephrology.
Conclusions
These complex physiological interactions with anatomical and physiological division of the nephron determine the stability and water and electrolyte balance of the body. Not only aquaresis, but also natriuresis and chloride serum concentration may constitute a practical and useful idea for the evaluation of efficient decongestion in patients with HF. Complex assessment, involving such elements as permeability of the proximal tubule wall and tubuloglomerular feedback, in the context of sodium and chloride balance, with the use of biomarkers of renal function, may play an important role in the evaluation of patients with HF at risk of AKI. Therefore, there is a need to update the concepts of CRS and strategies for their treatment in the context of renal function and its interaction with multiple organs.