Abstract
Background. Cervical cancer (CC) affects millions of women worldwide. This condition is strongly associated with human papillomavirus (HPV) infection. Oncogenic alterations are known to contribute to the development and progression of CC.
Objectives. This study aimed to screen for variations in selected genes associated with CC, including PIK3CA, KRAS, and PTEN, and to detect high-risk HPV genotypes 16, 18, 31, 45, 52, and 58 using gene-specific polymerase chain reaction (PCR), followed by single-strand conformation polymorphism (SSCP) analysis and confirmation using bidirectional DNA sequencing.
Materials and methods. The study included 414 participants, comprising 204 cases and 210 controls. Healthy controls were disease-free individuals participating in regular health checkups. Selected gene mutations were analyzed using PCR-based assays, SSCP, and sequence analysis. HPV genotyping was also performed.
Results. All study participants were analyzed for mutations in the PIK3CA, KRAS, and PTEN genes. The analysis revealed mutation frequencies of 6.37% for KRAS, 2.45% for PTEN, and 16.66% for PIK3CA in CC cases. These findings suggest that PIK3CA and KRAS mutations are more frequent in patients with CC than PTEN mutations. HPV infection was detected in 87.10% of patients with CC, 79.24% of participants with high-grade squamous intraepithelial lesions (HSIL), and 60.34% of participants with low-grade squamous intraepithelial lesions (LSIL). This study contributes to understanding the genetic basis of CC in South India and may facilitate the development of future targeted therapies.
Conclusions. The high prevalence of HPV underscores its etiological significance in CC. These findings contribute to a deeper understanding of the molecular mechanisms underlying CC in this population and may support the development of targeted therapeutic strategies for high-risk individuals. Future prospective studies and functional analyses are warranted to validate the clinical significance of these mutations and clarify their role in disease progression.
Keywords: genetics, cervical cancer, oncogenes, genotyping, mutations
Background
Cervical cancer (CC) is a major gynecological malignancy that arises in the lower part of the uterus, connecting the corpus uteri to the vagina.1 Despite advances in screening and vaccination programs, CC remains a significant public health burden, particularly in low- and middle-income countries.2 By 2026, an estimated 528,000 new cases of CC are expected, with developing nations contributing approx. 85% of the global burden.3 The disease accounts for about 266,000 deaths annually, representing 8% of all cancer-related mortality in women.4 In the USA alone, approx. 11,500 new cases are diagnosed each year, leading to 4,000 deaths.5 While CC incidence has declined in many developed countries due to effective screening programs,6 it remains the 4th most common malignancy among women worldwide, with 604,000 new cases and 342,000 deaths reported in 2020.7 India and China together account for over 1/3 of the global CC burden, with India reporting 97,000 new cases and 60,000 deaths annually. Notably, India has the highest age-standardized incidence rate in South Asia (22 per 100,000), with regional variations observed between South India (16.7–18.9 per 100,000) and the northeastern part of this country (24.3 per 100,000).8
Persistent infection with high-risk human papillomavirus (HPV) types is the primary cause of CC, contributing to more than 90% of cases.9 Among these, HPV 16 is the most oncogenic, followed by HPV 18.10 However, like many other malignancies, CC is not solely driven by viral infection but also by genetic alterations that dysregulate key signaling pathways. Mutations in oncogenes and tumor suppressor genes play a crucial role in tumor initiation and progression by disrupting cellular processes such as proliferation, metabolism, and apoptosis.11 Previous studies have identified genetic variations in multiple cancer-related genes, including AKT1, KRAS, HRAS, NRAS, PIK3CA, FGFR2, FGFR3, HER2, BRAF, EGFR, CCDC6, and PTEN, in patients with CC in China.12 However, research on the mutational profiles of PIK3CA, KRAS, and PTEN in South Indian patients with CC remains limited, leaving a gap in our understanding of the genetic landscape of this disease in this population.13
The PIK3CA gene, a key component of the phosphoinositide 3-kinase (PI3K) signaling pathway, is frequently mutated in various cancers, making it a potential biomarker for targeted therapies. The KRAS oncogene plays a critical role in tumorigenesis by regulating cell growth and proliferation, and its mutational status has clinical significance in multiple cancer types.14 The PTEN gene, a well-established tumor suppressor, regulates cell migration and inhibits uncontrolled tumor growth.15 Understanding the mutational status of these genes in CC could provide valuable insights into tumor biology and facilitate the development of more effective therapeutic strategies.
In regions with low socioeconomic status, the incidence and mortality of CC remain disproportionately high due to limited access to early diagnosis and personalized treatment.16 The limited availability of genetic profiling in South Indian patients with CC may complicate treatment decision-making and restrict opportunities for more individualized therapeutic approaches.17
Objectives
This study hypothesizes that mutations in PIK3CA, KRAS, and PTEN contribute to CC pathogenesis in South Indian women. The primary objective was to screen for genetic alterations in PIK3CA, KRAS, and PTEN, alongside high-risk HPV genotyping (types 16, 18, 31, 45, 52, and 58), and to assess their potential role in disease initiation and progression. By identifying specific mutations, this study aims to enhance understanding of the molecular mechanisms underlying CC in an understudied population and provide a foundation for future targeted therapeutic strategies.
Materials and methods
Sample collection
Samples for the study were collected from patients attending the Department of Obstetrics and Gynecology at Chettinad Hospital and Research Institute and Chettinad Super Speciality Hospital (CSSH) in Coimbatore, India. Patients were screened for HPV infection using type-specific polymerase chain reaction (PCR). The study protocol was approved by the Institutional Human Ethics Committee of the Chettinad Academy of Research and Education (CARE-IHEC; approval No. IHEC/D.NO:012), and informed consent was obtained from all participants prior to sample collection.
Inclusion and exclusion criteria
The study included patients diagnosed with CC across various histopathological grades, as well as healthy controls with no prior history of CC, HPV infection, or other gynecological disorders. Participants with a history of conization or hysterectomy, those who were pregnant, and individuals with severe comorbid conditions, sexually transmitted diseases, or other malignancies were excluded.
Sampling
Cervical scrapings were collected from study participants using a sterile disposable cervical cytobrush into a clean collection container (Citotest Labware Manufacturing Ltd., Haimen, China) for HPV detection and genetic analysis. Participants were also examined by trained gynecologists at Chettinad Hospital and Research Institute (Kelambakkam, India), who performed colposcopic examinations and obtained tissue samples when abnormalities were identified. The sample size was determined through power analysis using the formula n = p(1 − p)(Z/E)2, with a statistical power of 0.95.18 A total of 414 individuals were included, comprising 204 cases and 210 controls. The control group consisted of healthy individuals undergoing routine health checkups who were free of disease.
Extraction of genomic DNA
Genomic DNA was extracted from cervical scrapings using the phenol-chloroform method.19 DNA quantity and quality were assessed using spectrophotometric analysis, and gel electrophoresis was performed for further evaluation.
Mutation profiling and Sanger sequencing for mutation confirmation
According to the COSMIC database, previous studies have identified somatic mutations in PIK3CA, PTEN, and KRAS in cervical carcinoma.20 Therefore, these 3 genes were selected as targets for the present study, and gene-specific PCR primers were used to amplify mutation hotspot regions. Mutation detection within oncogenic hotspot regions was performed using gene-specific PCR.
Subsequent analyses included single-strand conformation polymorphism (SSCP) analysis and confirmation with bidirectional DNA sequencing. The nucleotide sequences of the exonic regions of PIK3CA, KRAS, and PTEN were retrieved from the Ensembl genome database (https://www.ensembl.org/index.html).21 Primers were newly designed for this study using the Primer3 (v. 4.0) program (https://primer3.ut.ee). The quality of the designed primers was evaluated using online tools, including Primer Stats and OligoCalc (https://www.bioinformatics.org/sms2/pcr_primer_stats.html and https://www.biosyn.com/gizmo/tools/oligo/oligonucleotide%20properties%20calculator.htm). The designed primer sequences and their properties are presented in Table 1. Polymerase chain reaction products were separated on a 1.2% agarose gel to visualize and analyze the specific amplicon bands. The amplified fragments were excised from the agarose gel and purified using the QIAquick PCR Purification Kit (cat. No. 28104) (Qiagen, Hilden, Germany). After purification, the DNA fragments were sequenced using the Sanger sequencing method22 with the Applied Biosystems 3130 system (Applied Biosystems, Foster City, USA). The obtained sequences were then compared with the reference sequences from the Ensembl genome database to identify mutations.
Statistical analyses
All statistical analyses were performed using IBM SPSS v. 21 (IBM Corp., Armonk, USA), with statistical significance set at p < 0.05. The normality of continuous variables, such as age, was assessed using the Shapiro–Wilk test, and appropriate parametric tests (Student’s t-test) were applied where assumptions of normality were met. Categorical variables were compared using Pearson’s χ2 test, while Fisher’s exact test was applied when expected cell counts were <5. For ordinal variables, trend analysis was conducted using the Cochran–Mantel–Haenszel (CMH) test.
Univariate risk ratios (RRs) with 95% confidence intervals (95% CIs) were calculated for selected demographic and lifestyle variables (e.g., tobacco use, contraceptive use, parity, and family history) to assess their association with CC risk. Multivariable logistic regression models with elastic net regularization (combining L1 and L2 penalties) were used to examine associations between mutations in PIK3CA, KRAS, and PTEN and clinical/lifestyle predictors, including HPV status, age, parity, contraceptive use, tobacco use, and family history of cancer. Ten-fold cross-validation was used to optimize the penalty parameters. Final model results are reported as adjusted odds ratios (aORs), 95% CIs, and p-values. Model classification performance was assessed using precision, recall, and F1 score at a probability threshold of 0.3. Receiver operating characteristic (ROC) curves and areas under the curve (AUCs) are presented in the supplementary materials.
Results
Clinical features of the cases and controls
The demographic and baseline characteristics of the study groups are presented as mean ± standard deviation (SD) in Table 2. The mean age of the participants was approximately normally distributed, as indicated by the Shapiro–Wilk test (p = 0.135 for cases; p = 0.838 for controls). As age was approximately normally distributed and differed modestly between groups, a 2-sample Student’s t-test was used for comparison. The analysis showed a significant difference between the case group (mean age: 53 ±8.90 years) and the control group (mean age: 50 ±5.49 years) (t = 4.14, degrees of freedom (df) = 412, p < 0.001).
Among the categorical variables, having more than 4 pregnancies (χ2 = 11.19, df = 1, p = 0.001), contraceptive use (χ2 = 9.93, df = 1, p = 0.002), family history of cancer (χ2 = 5.70, df = 1, p = 0.017), and tobacco use (χ2 = 7.38, df = 1, p = 0.007) were significantly more common in cases than in controls.
No significant differences between cases and controls were detected for education level (χ2 = 1.65, df = 3, p = 0.65), employment status (χ2 = 1.79, df = 2, p = 0.41), socioeconomic status (χ2 = 2.01, df = 2, p = 0.37), or the composite comorbidity category (χ2 = 2.28, df = 3, p = 0.52). In addition, risk ratios (RRs) with 95% CIs were calculated for binary variables. Having more than 4 pregnancies (RR = 2.14), contraceptive use (RR = 2.65), and tobacco use (RR = 2.27) showed strong positive associations with CC risk. Group differences in clinical and lifestyle factors were assessed using χ2 tests, and variables showing significant associations were considered potential confounders in the regression analysis. The normality of continuous variables, including age, was assessed using the Shapiro–Wilk test. The results are presented in Supplementary Table 1. All p-values in this section were derived from univariate analyses using Pearson’s χ2 test, where appropriate.
Genotyping of 6 high-risk HPVs
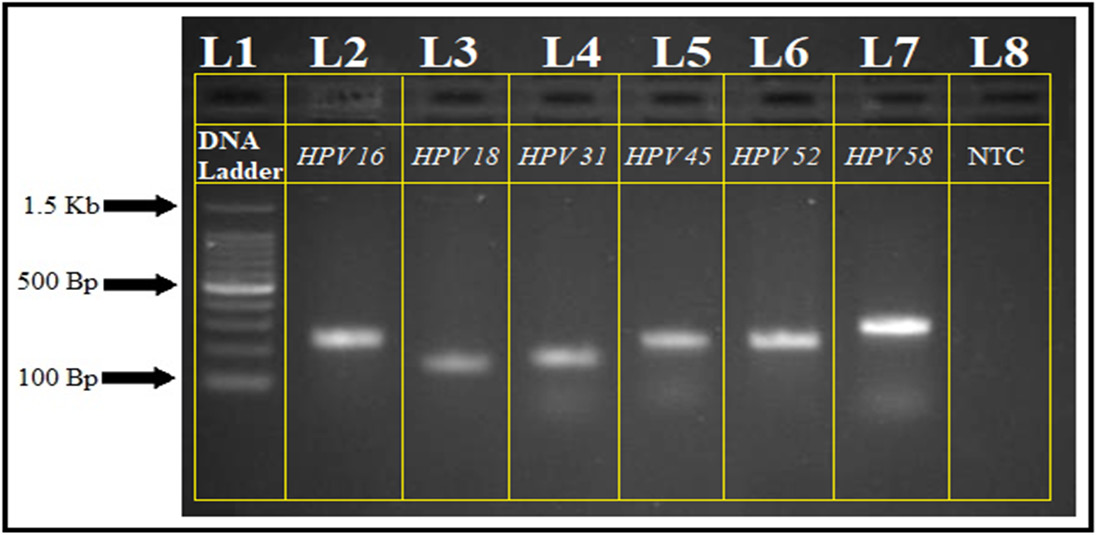
HPV genotyping was performed under optimized PCR conditions. Amplicons ranging from 150 to 295 base pairs indicated the presence of specific HPV genotypes in both cases and controls. Polymerase chain reaction products were analyzed using a 100 bp DNA ladder on a 1.2% agarose gel (Figure 1). The PCR findings were subsequently correlated with histopathological grading.
Pearson’s χ2 test revealed a highly significant association between HPV infection and histopathological grade (χ2 = 108.56, df = 3, p < 0.001), with HPV positivity increasing from negative for intraepithelial lesion or malignancy (NILM; 10.47%) to low-grade squamous intraepithelial lesion (LSIL; 60.34%), high-grade squamous intraepithelial lesion (HSIL; 79.24%), and CC (87.10%) (Table 3). A CMH test confirmed a significant increasing trend in HPV positivity with worsening histopathological grade (p for trend < 0.001). Among HPV genotypes, HPV 16 and HPV 58 showed significant variation across histopathological grades (HPV 16: χ2 = 8.10, df = 2, p = 0.017; HPV 58: χ2 = 9.11, df = 2, p = 0.033), with higher frequencies in more advanced lesions. No statistically significant distribution patterns were observed for HPV 18, 31, 45, or 52 (p > 0.05 for all) (Table 4).
Multivariable analysis of gene mutations
To assess the independent contribution of clinical and lifestyle factors to mutation risk, elastic net-regularized logistic regression was applied for PIK3CA and KRAS mutations. This method combines L1 and L2 penalties to balance variable selection and coefficient shrinkage and is particularly suitable for small or sparse datasets. The model identified CC status as the strongest independent predictor of both PIK3CA (aOR = 74.03) and KRAS (aOR = 7.44) mutations. Other variables, including HPV positivity, tobacco use, parity, contraceptive use, and family history of cancer, were retained in the models but did not demonstrate statistically significant associations (Table 5).
Mutation profiling
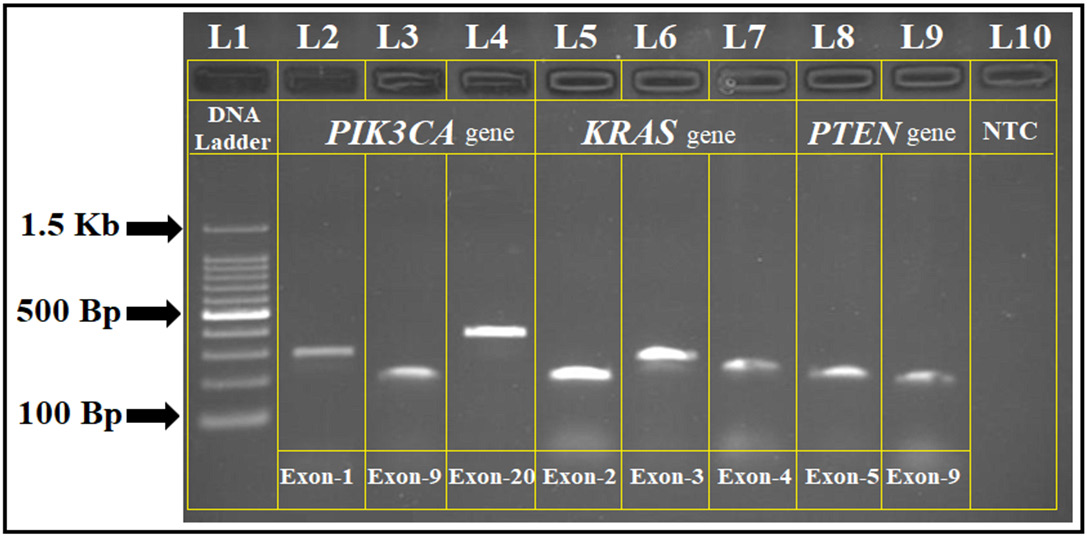
Mutations in the PIK3CA, KRAS, and PTEN genes were analyzed among study participants using gene-specific PCR. The amplicon sizes ranged from 180 to 400 base pairs, and the PCR products were confirmed with agarose gel electrophoresis (Figure 2). Mutation frequency comparisons between cases and controls were assessed using univariate ORs.
The observed mutation frequencies among CC cases were 16.66% for PIK3CA (95% CI: 12.34–20.98, p = 0.002), 6.37% for KRAS (95% CI: 4.11–8.63, p = 0.017), and 2.45% for PTEN (95% CI: 1.12–3.78, p = 0.042). No significant mutations were identified in the control group, resulting in perfect separation for some outcomes. Statistical comparisons between cases and controls confirmed a strong association between PIK3CA mutations and CC risk (OR = 3.89, 95% CI: 2.14–7.08, p < 0.001).
Gene profiling of PIK3CA, KRAS, and PTEN
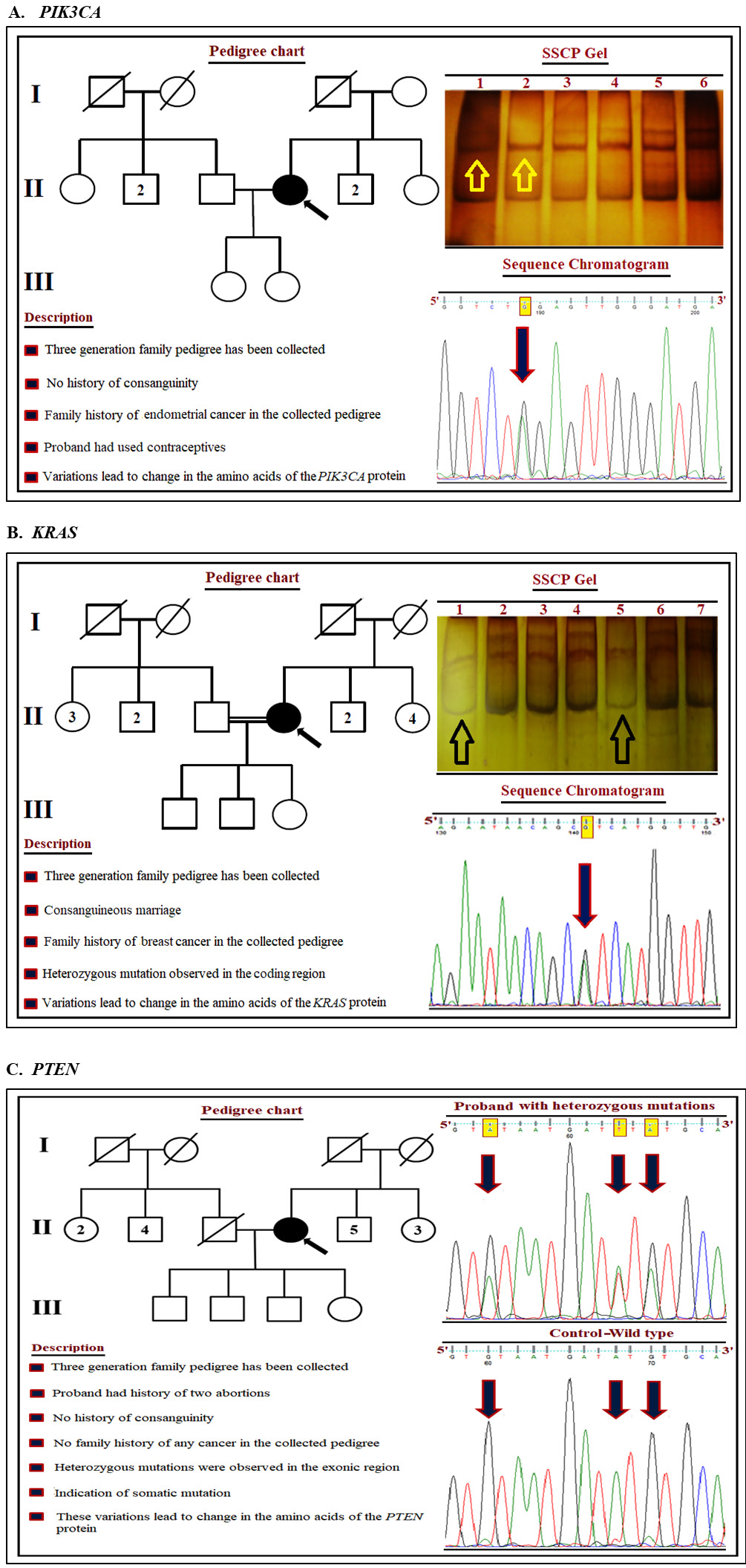
Multivariable logistic regression was performed to assess the association between gene mutations and potential confounding factors. Penalized (L2) logistic regression was applied due to sparse mutation events and perfect separation in some models. Results are presented as aORs with 95% CIs. The distribution of mutations in PIK3CA, KRAS, and PTEN was further analyzed in relation to HPV genotype and histopathological grade (Figure 3). PIK3CA mutations were predominantly identified in exons 9, 20, and 1, with hotspot mutations located in the helical domain (E542K, E545K) and catalytic domain (H1047R). The overall frequency of these mutations was 2.94%. Patients harboring PIK3CA mutations had a significantly higher likelihood of HPV 16 infection (p = 0.004). Detailed information regarding PIK3CA mutations in relation to HPV genotypes and histopathological grading is presented in Table 6. KRAS mutations were primarily detected in exon 2, followed by exons 3 and 4 (Table 7). These mutations were significantly associated with a family history of breast cancer and consanguineous marriage (p = 0.016). As shown in Table 8, logistic regression analysis demonstrated that KRAS mutations were significantly associated with HPV positivity (OR = 2.47, 95% CI: 1.32–4.21, p = 0.022) and CC (OR = 3.80, 95% CI: 2.01–6.88, p < 0.001). An interaction term between HPV status and cancer status was also statistically significant.
PTEN gene variants were predominantly identified in exon 5, followed by exon 9. These mutations were detected exclusively in CC cases and were not observed in any control subjects. Patients harboring PTEN mutations did not report a family history of cancer or consanguinity, suggesting that these variants may represent somatic alterations contributing to carcinogenesis. A novel PTEN mutation identified in this study has not previously been reported in HPV-associated CC (Table 9).
Model performance evaluation
To further evaluate the classification performance of the mutation prediction models, various probability thresholds (e.g., 0.3) were tested. However, due to extreme class imbalance, the models failed to identify mutation-positive cases (zero true positives). Despite this limitation, overall accuracy remained relatively high (91.8% for PIK3CA and 96.9% for KRAS), largely driven by the high number of correctly classified negative cases. These results are presented in Supplementary Table 2. Nevertheless, ROC analysis indicated that both models retained moderate-to-strong discriminatory ability, with AUC values of 0.825 for PIK3CA and 0.881 for KRAS (Supplementary Table 3). The corresponding ROC curves are shown in Supplementary Fig. 4.
Discussion
Advances in high-throughput sequencing technologies have enabled the identification of novel genetic variants and somatic mutations with high depth of coverage, contributing to the transition toward precision medicine. The fundamental principle of mutation profiling is the detection of genetic alterations in candidate or disease-associated genes. This approach may facilitate the identification of predictive biomarkers and improve the effectiveness of therapeutic strategies.
The present study aimed to identify and characterize CC-associated mutations using gene-specific PCR, followed by SSCP analysis and confirmation by DNA sequencing. According to our findings, mutations in PIK3CA (16.66%), KRAS (6.37%), and PTEN (2.45%) were associated with CC in South Indian patients. PIK3CA is one of the genes frequently implicated in human malignancies. The PI3K signaling pathway plays a critical role in multiple cellular processes, including cell survival, metabolism, growth, and proliferation. PIK3CA mutation and amplification are among the most common mechanisms leading to aberrant activation of the PI3K pathway in various cancers.23 The mutation frequency of PIK3CA in our study was 16.66% among patients with CC. When compared with previous studies, a cohort from the Netherlands including 301 patients with CC of Caucasian ancestry reported PIK3CA mutations in 20% of cases. Similarly, a study of 213 Chinese patients with CC reported a mutation frequency of 12.3%.24 Another study involving Chinese patients of Asian ancestry that screened 16 genes also reported a PIK3CA mutation frequency of 12.3%.25 Our observed PIK3CA mutation frequency was therefore somewhat higher than that reported in Chinese cohorts, despite both populations being of Asian ancestry. These differences may reflect variation in mutation spectra, patient characteristics, HPV subtype distribution, or methodological differences between studies. A study from the USA involving 67 patients with CC reported PIK3CA mutations in 27.1% of cases, which is substantially higher than the frequency observed in our cohort.26 The higher prevalence of PIK3CA mutations in some Western populations may reflect differences in environmental exposures, lifestyle factors, genetic background, or interactions with high-risk HPV variants.
The KRAS gene is a well-established oncogene, and multiple point mutations have been identified across various malignancies, including breast, cervical, endometrial, liver, and myeloid cancers.27 The KRAS protein plays a crucial role in regulating the MAPK signaling pathway and may also influence cellular proliferation through interactions with the PI3K–AKT pathway. Our study demonstrated an association between KRAS mutations and high-risk HPV 16/18 genotypes. The integration of KRAS mutational analysis with HPV genotyping may improve risk stratification and potentially inform prognostic assessment in patients with CC. The mutation frequency of KRAS in this study was 6.37% among patients with CC. In comparison, a previous study from Boston analyzing 80 Caucasian patients across 139 cancer-related genes reported an overall mutation rate of 60%, with KRAS mutations accounting for 8.8% of cases.28 In a Chinese study (n = 876), KRAS mutations were detected in 3.4% of CC cases,29 whereas another Chinese study focused on cervical adenocarcinoma reported KRAS mutations in 16.6% of patients.30 These differences may reflect variation in histological subtype, study population, sequencing methodology, or sample composition.
Although KRAS mutations have been reported across diverse populations, current evidence remains insufficient to conclude that they represent a conserved oncogenic event in CC irrespective of ethnicity. In addition to mutation-driven signaling pathways, targeted gene-silencing approaches such as MCT1 inhibition have shown promise in enhancing dendritic cell-mediated immune responses against CC, suggesting potential synergy between immunotherapeutic and molecular targeting strategies.31 However, our observed association between KRAS mutations and HPV 16/18 infection highlights the potential value of integrating viral and genetic profiling in CC risk assessment.
PTEN is a well-established tumor suppressor gene implicated in the development of multiple cancer types and plays an important role in regulating cellular proliferation, survival, and migration. The phosphatase encoded by PTEN negatively regulates the PI3K signaling pathway through dephosphorylation of phosphoinositide substrates, thereby limiting uncontrolled cellular growth and proliferation.32 In our study, the mutation frequency of PTEN was 2.45% among patients with CC. Compared with previous studies, cohorts from Mexico and Norway analyzing 115 CC and control samples reported a PTEN mutation frequency of approx. 6%, which is higher than the frequency observed in our cohort.33 A Japanese study (n = 50) reported PTEN mutations in 4.2% of cases,34 while a study from the USA reported PTEN alterations in 8% of CC cases.35 Another study in Mexican patients (n = 155) identified PTEN mutations in 5% of cases.36
Observed differences in PTEN mutation frequency across studies may reflect variation in study population characteristics, sequencing methodology, histological subtype composition, or sample size rather than ethnicity alone. The reasons for these discrepancies remain uncertain. In addition, the identification of a novel PTEN mutation in our study raises the possibility of previously unrecognized genetic alterations in South Indian patients with CC, warranting further investigation.
Limitations of the study
One important limitation of this study is its focus on a specific population from South India, which may limit the generalizability of the findings to other populations or ethnic groups. Additionally, the relatively small sample size, particularly for mutation frequency analyses, may have reduced the statistical power to detect less common genetic variants. Although techniques such as SSCP and bidirectional DNA sequencing were employed, these methods may not detect all potentially relevant genetic alterations, including large structural variants or epigenetic changes. Furthermore, the use of conventional PCR and Sanger sequencing rather than next-generation sequencing (NGS) may have limited the ability to detect rare, novel, or non-hotspot mutations more comprehensively. A major limitation of the mutation prediction models was their inability to accurately identify mutation-positive cases because of the low frequency of mutation events and severe class imbalance. This resulted in zero sensitivity and F1 scores at the evaluated classification thresholds. Therefore, further large-scale studies using more comprehensive genomic approaches are needed to better understand the genetic landscape of CC in South Indian patients and its relationship to other populations.
Conclusions
Our findings underscore the importance of understanding population-specific genetic variation in CC. The higher mutation frequency of PIK3CA compared with KRAS and PTEN suggests that alterations in the PI3K pathway may play a prominent role in cervical carcinogenesis in South Indian women. These results provide valuable insights into the molecular landscape of CC in this population and add to the growing body of evidence suggesting that genetic alterations, together with HPV infection, may influence disease progression and therapeutic response. This study highlights the potential relevance of PIK3CA, KRAS, and PTEN mutations in CC among South Indian women. A higher mutation frequency was observed for PIK3CA (16.66%) compared with KRAS (6.37%) and PTEN (2.45%), suggesting that alterations in the PI3K/AKT/mTOR pathway may play an important role in cervical carcinogenesis in this population. The observed association between KRAS mutations and high-risk HPV 16/18 infection warrants further investigation into the biological and potential clinical significance of this finding. Although PTEN mutations were less frequent, their established role in tumor suppression suggests potential relevance to disease biology. Integrating genetic profiling with HPV testing may improve molecular characterization and risk stratification in CC. However, the clinical utility of mutation-guided therapeutic strategies, including PI3K-targeted therapies, alternative approaches for KRAS-mutant tumors, or the predictive relevance of PTEN alterations for immunotherapy response, requires further validation in larger prospective studies. Future research should focus on further investigating these mutations and clarifying their potential clinical relevance. Our findings suggest that more individualized therapeutic strategies for CC warrant exploration. Although current management strategies do not routinely incorporate molecular stratification beyond standard clinicopathological classification, our study suggests that identifying distinct molecular subpopulations within CC may offer opportunities to improve patient stratification and potentially enhance outcomes in both early- and advanced-stage disease.
Supplementary data
The supplementary materials are available at https://doi.org/10.5281/zenodo.16919759. The package contains the following files:
Supplementary Table 1. Normality test results for continuous variables.
Supplementary Table 2. Performance metrics and confusion matrix for elastic net logistic regression models.
Supplementary Table 3. Performance metrics for elastic net logistic regression models.
Data Availability Statement
Data sharing does not apply to this article, as all data are already included in the manuscript.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.