Abstract
Background. Radiotherapy is the main treatment for nasopharyngeal carcinoma. The radioresistance mechanism of cells is related to miRNAs.
Objectives. To investigate the miRNA profiling of HONE1 and CNE2 after X-ray therapy.
Materials and methods. The HONE1 and CNE2 cells were treated with X-ray at 4 Gy, 8 Gy, 16 Gy, and 20 Gy doses. The cell lines CNE2 with the best therapy effects and HONE1 with the worst therapy effects were screened out. Apoptosis and cell viability were detected with flow cytometry and Cell Counting Kit-8 (CCK-8). High-throughput sequencing was performed. A miRNA library was constructed. The miRNA annotation expression distribution, family prediction and target gene interaction, Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were conducted.
Results. The 24-hour 20 Gy dose X-rays were selected as the optimal therapy conditions. The CNE2_C, CNE2_M, HONE1_C and HONE1_M miRNAs accounted for 26.5%, 31.7%, 21.3%, and 22.9% of the Cleandata reads count, respectively, and the contents of rRNAs accounted for 24.9%, 14.7%, 25.1%, and 25.1% of the Cleandata reads count, respectively. The miRNAs with differential expression between the HONE1 and CNE2 cell lines including hsa-miR-21-5p, hsa-let-7a-5p, hsa-miR-125a-5p, hsa-miR-26a-5p, hsa-let-7f-5p, hsa-miR-20a-5p, and hsa-miR-24a-3p. There were also differentially expressed miRNAs in HONE1_C vs. HONE1_M, such as hsa-miR-21-5p and hsa-let-7i-5p. The differentially expressed miRNA in CNE2_C vs. CNE2_M was hsa-miR-148b-3p. The Gene Ontology analysis showed that the differentially expressed miRNA interacting genes in HONE1_M vs. CNE2_M were mainly enriched in biological process such as negative and positive regulation of transcription from RNA polymerase II promoter, cellular component such as cytosol and molecular function such as protein binding factor. The KEGG pathway analysis revealed that the differentially expressed miRNA interacting genes in HONE1_M vs. CNE2_M were enriched in the cancer-related pathways, such as pathways in cancer, MAPK signaling pathway and Wnt signaling pathway.
Conclusions. Twelve miRNAs and 9 genes which contribute to X-ray radiation resistance were identified. Among those with differential expression between the HONE1 and CNE2 cell lines, which played a regulatory role in multiple pathways, were hsa-miR-20a-5p, hsa-let-7a-5p, hsa-let-7f- 5p, hsa-let-7i-5p, hsa-miR-30e-5p, hsa-miR-148b-3p, and hsa-miR-200c-3p. The corresponding genes were MAPK1, SOS1, TGFBR1, TGFBR2, TP53, CASP3, CCNE2, PTEN, and CDK2.
Key words: HONE1, CNE2, X-ray radiation resistance, human nasopharyngeal carcinoma, miRNA profiling
Background
Nasopharyngeal carcinoma (NPC) is a type of squamous cell carcinoma formed in nasopharyngeal tissue.1 It is mainly divided into poorly differentiated squamous cell carcinoma and undifferentiated carcinoma, of which type III NPC is the most common subtype.2 The NPC can be induced by long-term smoking, Epstein–Barr virus (EBV) infection and genetic factors.3, 4 The incidence of NPC is regional, and mainly focused in the southern part of China, Southeast Asia and Northern Africa.5, 6
The main treatment for head and neck cancer is surgery. However, due to the specific anatomical structure and growth characteristics of NPC, it is not suitable for surgical treatment.7 Therefore, radiotherapy is the main treatment for NPC. Radiotherapy can directly kill the cancer cells or destroy the DNA structure of the cancer cells, rendering the cancer unable to multiply indefinitely and thus achieving the purpose of treatment. However, radiotherapy has certain limitations. Due to the different cell cycle of cancer cells, the sensitivity to radiotherapy is also different. Cells in the vigorous division stage are more sensitive, and cells in the stable stage have certain resistance to radiotherapy.7
Studies have found that the radioresistance mechanism of cells is related to the regulation of apoptosis, proliferation and migration of the cancer cells by miRNAs.8, 9 The miRNA coding genes are located in the introns of the protein coding genes and the introns or exons of noncoding genes.10 The miRNAs can be fully or partially complementary to the 3’-UTR sequence of mRNA, and inhibit mRNA expression by cutting mRNA or repressing mRNA translation.11 Usually, a single miRNA can regulate multiple genes, or multiple miRNAs can regulate one gene. Radiotherapy for NPC does not guarantee curing the disease. There is a large number of people experiencing relapse who need to be treated again.12 The miRNA-based therapeutic approach has the benefit of being able to target multiple effectors of pathways participating in tumor cell differentiation and proliferation concurrently. Onco-miRNAs, tumor suppressive miRNAs, metastasis promoter or suppressor miRNAs, and EBV-encoded miRNAs could be a useful therapeutic strategy for NPC. Exosomal miRNAs might also serve as useful indicators of NPC.13
Research on the resistance mechanism of cancer cells to radiotherapy plays an important role in the treatment of NPC. With the rapid development of high-throughput sequencing in recent years, we can detect the type and expression of miRNA in cancer tissue, and use bioinformatics analysis to predict miRNA-targeted regulation genes and action pathway, so as to lay the foundation for future research on the mechanism of radiotherapy resistance of NPC.
Objectives
In this study, we investigated the miRNA profiling of HONE1 and CNE2 after X-ray therapy. We subjected the X-ray-sensitive NPC cell line CNE2 and X-ray-resistant NPC cell line HONE1 screened in the preliminary experiment to radiotherapy, performed post-treatment miRNA high-throughput sequencing for the control and model groups, miRNA species and expression analysis, target gene prediction and regulatory pathway enrichment analysis, and compared the differences of miRNAs in the 2 types of cell lines. The findings of this study may provide a reference for future research on anti-X-ray mechanism in NPC cells.
Materials and methods
Experimental materials
The HONE1 cells (BNCC338405) and CNE2 cells (BNCC100088) were purchased from Bena Culture Collection (Beijing, China).
This study was conducted in strict accordance to the standard guidelines and university procedures.
Experimental reagents and instruments
The following reagents and equipment were used: Qubit 2.0 RNA detection kit (Q32855) and Qubit 2.0 DNA detection kit (Q10212; Life Technologies Corp., Carlsbad, USA); T4 RNA Ligase 1 (0011309) and T4 RNA Ligase 2 (0511412; New England Biolabs, Ipswich, USA); M-MuLV Reverse Transcriptase (B600005; Sangon Biotech Co., Ltd., Shanghai, China); Qubit2.0 fluorometer (Q32866; Invitrogen, Waltham, USA); micro vortex mixer (WH-3; Shanghai Luxi Analysis Instrument Factory Co., Ltd., Shanghai, China); desktop high-speed low-temperature centrifuge (Thermo Scientific Sorvall Legend Micro 21R; Thermo Fisher Scientific, Waltham, USA); polymerase chain reaction (PCR) instrument (T100TM Thermal Cycler; Bio-Rad Laboratories, Inc., Hercules, USA); electrophoresis instrument (DYY-11; Beijing Liuyi Biotechnology Co., Ltd., Beijing, China); and imaging analysis of nucleic acid and protein (FR-980A; Shanghai Furi Science & Technology Co., Ltd., Shanghai, China).
Cell culture
The cells were plated, washed twice with 1× phosphate-buffered saline (PBS) and digested with 0.25% trypsin (containing 0.02% ethylenediaminetetraacetic acid (EDTA)) for 2–3 min; then, medium was added to terminate digestion and the cells were suspended. Then, the cells were collected into a 10-mL centrifuge tube, centrifuged at 1000 rpm for 3 min and the supernatant was discarded. Culture medium was added and the cell suspension was blown evenly through a pipette. After cell counting, the above cell suspension was diluted into the appropriate cell density according to the grouping, added to the prepared culture plate and cultured in a 37°C 5% CO2 incubator.
X-ray irradiation
When the cells were inoculated to 70% confluence, the model was initiated and 2 types of cells (HONE1 and CNE2) were treated with X-rays at 4 Gy, 8 Gy, 16 Gy, and 20 Gy doses. After treatment, flow cytometry apoptosis detection and Cell Counting Kit-8 (CCK-8) detection were performed.
Apoptosis detected with flow cytometry
Approximately 1 × 106 to 3 × 106 cells were collected, 1 mL of PBS was added, and then the cells were centrifuged at 1500 rpm for 3 min and washed twice. Double distilled water was used to dilute the 5× binding buffer to 1× binding buffer, and 300 μL of precooled 1× binding buffer was used to resuspend the cells. Then, 3 μL of Annexin V-APC and 5 μL of 7-AAD were added into each tube, slightly mixed, and incubated at room temperature (RT) in the dark for 10 min. Subsequently, 200 μL of precooled 1× binding buffer was added into each tube, mixed and detected using flow cytometry.
Cell viability detected by CCK-8
After X-ray irradiation of HONE1 and CNE2 cells, 10 μL of CCK-8 detection reagent were added into each well of a 96-well plate and incubated for 2 h at 37°C. The optical density (OD) value of each well was detected at 450 nm wavelength with the microplate reader and the cell viability rate was calculated.
Construction of miRNA library
According to the small RNA with 3’-hydroxyl group and 5’-phosphate group structural characteristics, 3’ adaptor ligation, reverse transcription primer hybridization, 5’ adaptor ligation, cDNA single-stranded synthesis, and library amplification were performed on 4 types of cell samples – HONE1_Control (HONE1_C), HONE1_Model (HONE1_M), CNE2_Control (CNE2_C) and CNE2_Model (CNE2_M) – using related enzyme catalytic reaction characteristics and molecular biology techniques. After quality inspection and purification, a library that satisfied the sequencing requirement of the Illumina platform (http://www.usadellab.org/cms/?page=trimmomatic) was finally obtained.
Database and analysis methods
The databases used in the study included:
1. Rfam (v. 12.0, http://rfam.xfam.org/)
Rfam is a database of the non-coding RNA (ncRNA) family expressed by multiple sequence alignment and covariance model (CM) for homology detection and sequence alignment, and used for enrichment analysis of the miRNA family.
2. miRBase (v. 21; http://www.mirbase.org/ftp.shtml)
After miRNA quantification, the analysis was performed on miRBase with known miRNA family information.
3. miRDB (http://mirdb.org/index.html), miRTarBase 7.0 (http://www.microrna.gr/tarbase)
This database is used to predict miRNAs and target binding genes.
4. mirPath (http://www.microrna.gr/miRPathv2)
This database is used to integrate miRNA interacting genes into various regulatory pathways.
5. Gene Ontology (GO; http://www.geneontology.org/)
The GO database is used for enrichment analysis of gene function.
6. Kyoto Encyclopedia of Genes and Genomes (KEGG pathway; http://www.genome.jp/kegg/pathway.html)
This database is used to enrich and analyze the role of miRNA in various interaction pathways.
The miRNA quality control analysis
The 3’ adapter was removed from the raw data. The software used was Cutadapt (https://cutadapt.readthedocs.io/
en/stable/installation.html). The length of reads after removing the adapter was set to be within 17–35 bp. The reads after removing the adapter were treated with Trimmomatic (v. 0.36; http://www.usadellab.org/cms/?page=
trimmomatic).14 The bases with a quality value lower than 20 at the 5’ and 3’ terminals were deleted; 4 consecutive bases with an average quality value of <20 and reads with length of <17 after processing were filtered out in order to obtain total reads. The reads with duplicates removed were called unique reads and the number of reads was statistically analyzed.
Comparison of reference genomes and classification annotation
Bowtie (http://bowtie-bio.sourceforge.net/index.shtml) was used to compare the reads of each sample with the reference genome (mismatch was set to ≤1). The number and percentage of reads were counted, and the reads that could not be compared to the reference genome were filtered. The reads were annotated and counted in the sequence of rRNA, tRNA, snRNA, snoRNA, miRNA, and others. The reads of miRNA sequencing might contain other small RNAs. The analysis was performed using Blastn search tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi) for reads, with rRNA, sRNA, snRNA, and snoRNA of the Rfam2 database. The alignment conditions were set as follows: gapopen 0, evalue < 0.01 and mismatch ≤ 1, and the reads on the alignment were filtered out.
The miRNA family prediction
MicroRNA family is a group of miRNAs from the same ancestor. They usually have similar biological functions, but not necessarily conservative in primary and secondary structures. After miRNA quantification was completed, the known miRNA family information on miRBase was used for analysis. The miRNAs that did not belong to any family were represented by “not applicable” (N/A). These miRNAs is not classified into any miRNA family; some detected miRNAs have not been studied yet, and their structures are different from the miRNA structures of the currently known families.
The miRNA expression analysis
The expression levels of miRNAs were evaluated. By calculating reads per million (RPM), the counts were normalized to RPM value. The boxplot was constructed for the RPM of the sample to show the RPM distribution. This article mainly displayed the data analysis of the top 20 miRNAs with the highest total miRNA expressions.
The miRNA target gene prediction
The miRTarBase (http://www.targetscan.org/vert_72/) was used to predict the target genes that the miRNAs might bind to in the cancer gene pathway. The miRDB database was used to rescreen genes that might interact with miRNAs. TargetScan database is a predictor that produces predicted interactions, while miRTarBase and miRDB databases provide verified target genes that have binding sites with miRNAs. The miRNAs and target genes that play a role in cancer genes were screened out and the miRNA-gene interaction network was constructed using Cytoscape v. 3.8.0 (https://cytoscape.org/).
Gene enrichment analysis
The mRNA-targeted binding genes were used as the target gene. The GO and KEGG pathway annotation of the species were used for enrichment analysis. By classifying, sorting and analyzing GO functional annotations, the differences in the GO annotation distribution of different target gene sets were displayed in the form of a bar graph, and the biological significance of corresponding miRNAs was explored. The software used for GO enrichment analysis was clusterProfiler (https://www.rdocumentation.org/packages/clusterProfiler
/versions/3.0.4), and the network diagram drawing software was iGraph (https://igraph.org/). The KEGG pathway database collects biological pathways. Each pathway contains molecular interactions and metabolic reactions, a network diagram connecting genes and gene products. The clusterProfiler was used for KEGG pathway enrichment analysis.
Statistical analyses
All bioinformatics statistics were performed using R software (https://www.r-project.org). The data obtained from the cells experiment were plotted using GraphPad Prism v. 8.0.1 (GraphPad Software, San Diego, USA), and analyzed with IBM SPSS v. 19.0 (IBM Corp., Armonk, USA). The comparison between groups was performed using Kruskal–Wallis test. A value of p < 0.05 indicated significant difference.
Results
Cellular X-ray therapy modeling
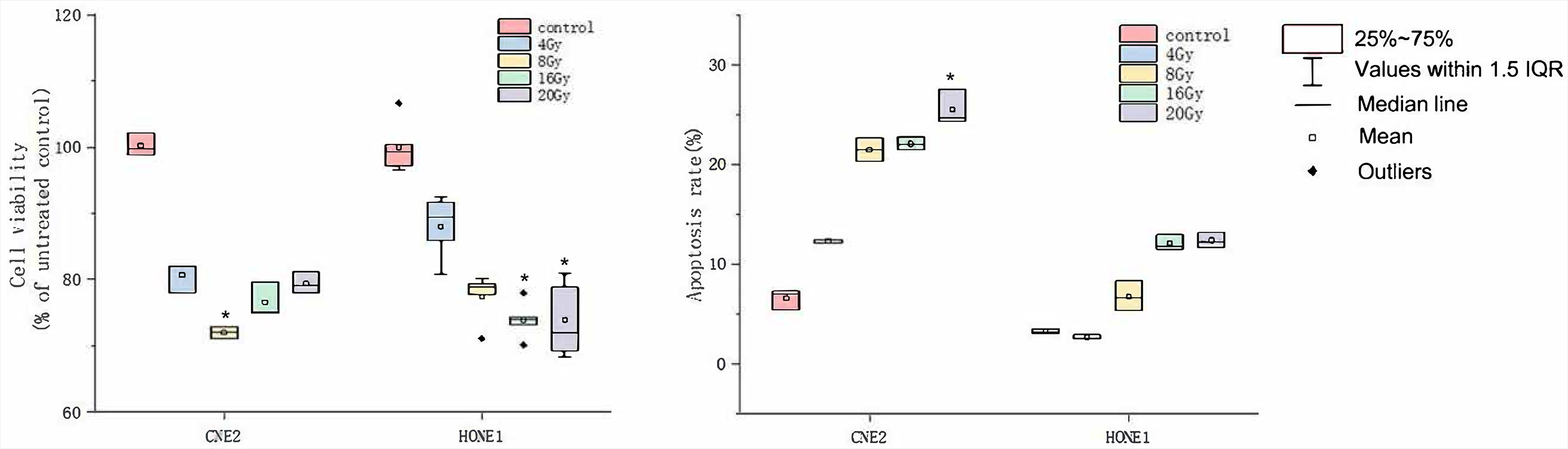
In the preliminary experiment, we irradiated several NPC cell lines with different doses of X-rays, established a 24-hour 20 Gy X-ray dose as the optimal therapy conditions, and screened out the NPC cell line CNE2 with the best therapy effects and NPC cell line HONE1 with the worst therapy effects. The CCK-8 and flow cytometry detection of cell viability and apoptosis were performed on these 2 cell lines. The HONE1 and CNE2 cells were irradiated with X-rays at doses of 4 Gy, 8 Gy, 16 Gy, and 20 Gy for 24 h. Compared with the control group, the cell viability rate for CNE2 at 8 Gy dose and for HONE1 at 16 Gy and 20 Gy doses decreased significantly; the apoptosis rate for CNE2 at 20 Gy dose increased significantly (Figure 1 and Supplementary Table 1 and 2: https://doi.org/10.5281/zenodo.6132842). High-throughput sequencing was performed on the modeled CNE2_C, CNE2_M, HONE1_C, and HONE1_M groups.
miRNA quality control analysis
The quality of the sequenced raw data was evaluated using FastQC (https://www.bioinformatics.babraham.ac.uk/
projects/fastqc/), and the Rawdata reads were screened. The screening condition was set to reads, with a length of 17–35 bp, and an average base quality value >20. The Cleandata reads were obtained. After removing the duplicate data, Uniq_reads were acquired. Compared with the number of CNE2_C miRNA reads, the number of miRNA reads in the CNE2_M group was significantly increased. The results are shown in Table 1.
The miRNA annotation analysis
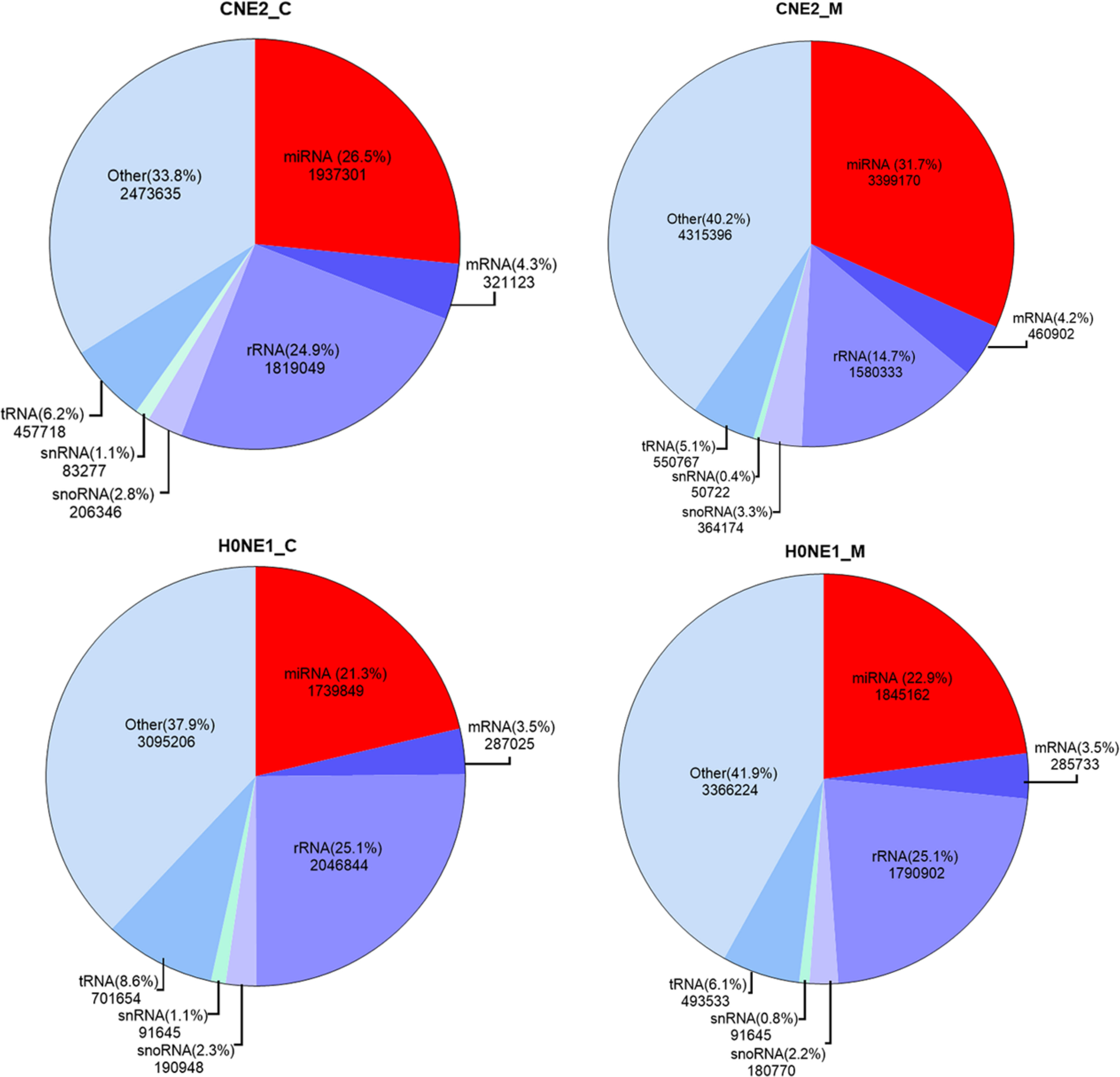
Blastn search tool was used to compare the reads in CNE2_C, CNE2_M, HONE1_C and HONE1_M with tRNA, snRNA, snoRNA, rRNA, mRNA, and miRNA in the Rfam database. The number and percentage of reads of the comparison were calculated, and a pie chart was built (Figure 2). The miRNAs accounted for 26.5%, 31.7%, 21.3%, and 22.9% of the Cleandata reads count in CNE2_C, CNE2_M, HONE1_C and HONE1_M, respectively. The contents of tRNA, snRNA, snoRNA, rRNA, and mRNA are shown in Figure 2. The contents of rRNA were relatively high, with 24.9%, 14.7%, 25.1%, and 25.1% of the Cleandata reads count, respectively.
The miRNA expression distribution analysis
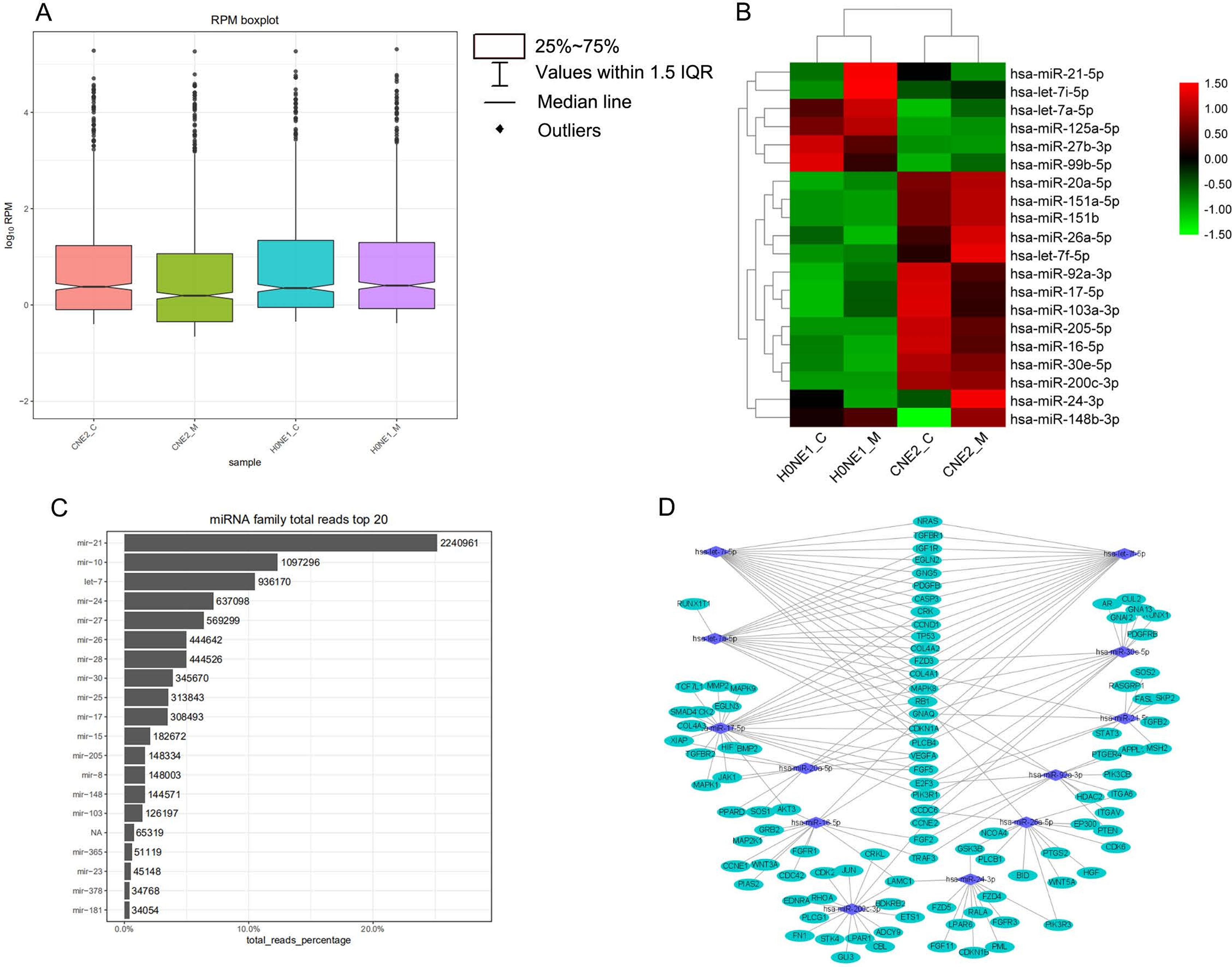
In order to show the overall expression distribution of miRNAs in each group, the RPM log10 of miRNAs in each group was taken and a box plot was built, as shown in Figure 3A. It can be seen that the median of the CNE2_M group was lower than in the other groups, indicating that in the CNE2_M group, there were more miRNAs with low expression levels. However, in the quality control analysis, the miRNA_total_reads of the CNE2_M group was higher than of the other 3 groups, indicating that the expression levels of some miRNAs in the CNE2_M group was abnormally high. We took the top 20 miRNAs in expression for heat map cluster analysis, as shown in Figure 3B. The Z-cord value of RPM was used for the heat map cluster analysis. Red represented high expression and green represented low expression. It could be seen that in the CNE2_M group, the amount of miRNAs with expressions which appeared red was higher than in the other 3 groups. The expressions of the HONE1 cells were low and appeared as green miRNAs, while most of those in the CNE2 group were relatively high and appeared red. Among them, there were significant differences in the expressions of hsa-miR-21-5p, hsa-let-7a-5p, hsa-miR-125a-5p, hsa-miR-26a-5p, hsa-let-7f-5p, hsa-miR-20a-5p, and hsa-miR-24a-3p between the HONE1 and CNE2 cell lines. There were also differentially expressed miRNAs in HONE1_C vs. HONE1_M, such as hsa-miR-21-5p and hsa-let-7i-5p, and in CNE2_C vs. CNE2_M, such as hsa-miR-148b-3p.
The miRNA family prediction analysis
The quantified miRNAs were analyzed using the known miRNA family information in the miRBase, and miRNAs that did not belong to any family were marked as N/A. We selected the top 20 miRNAs in family total reads for family prediction analysis. As shown in Figure 3C, these families were mir-21, mir-10, let-7, mir-24, mir-27, mir-26, mir-28, mir-30, mir-25, mir-17, mir-15, mir-205, mir-8, mir-148, mir-103, mir-365, mir-23, mir-378, and mir-181. Among them, the reads of miR-21, mir-10 and let-7 families were relatively higher than reads for other miRs.
The miRNA target gene interaction analysis
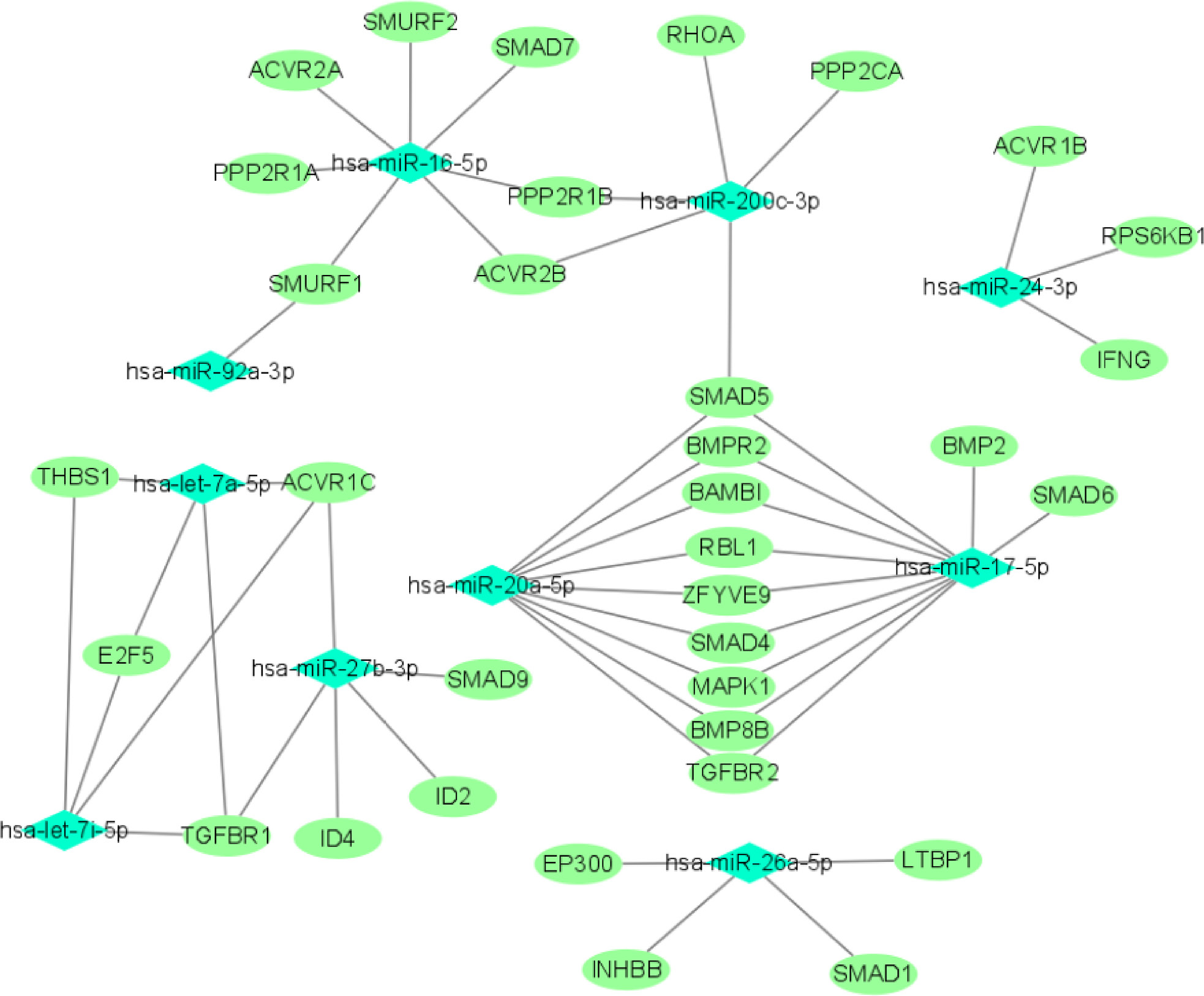
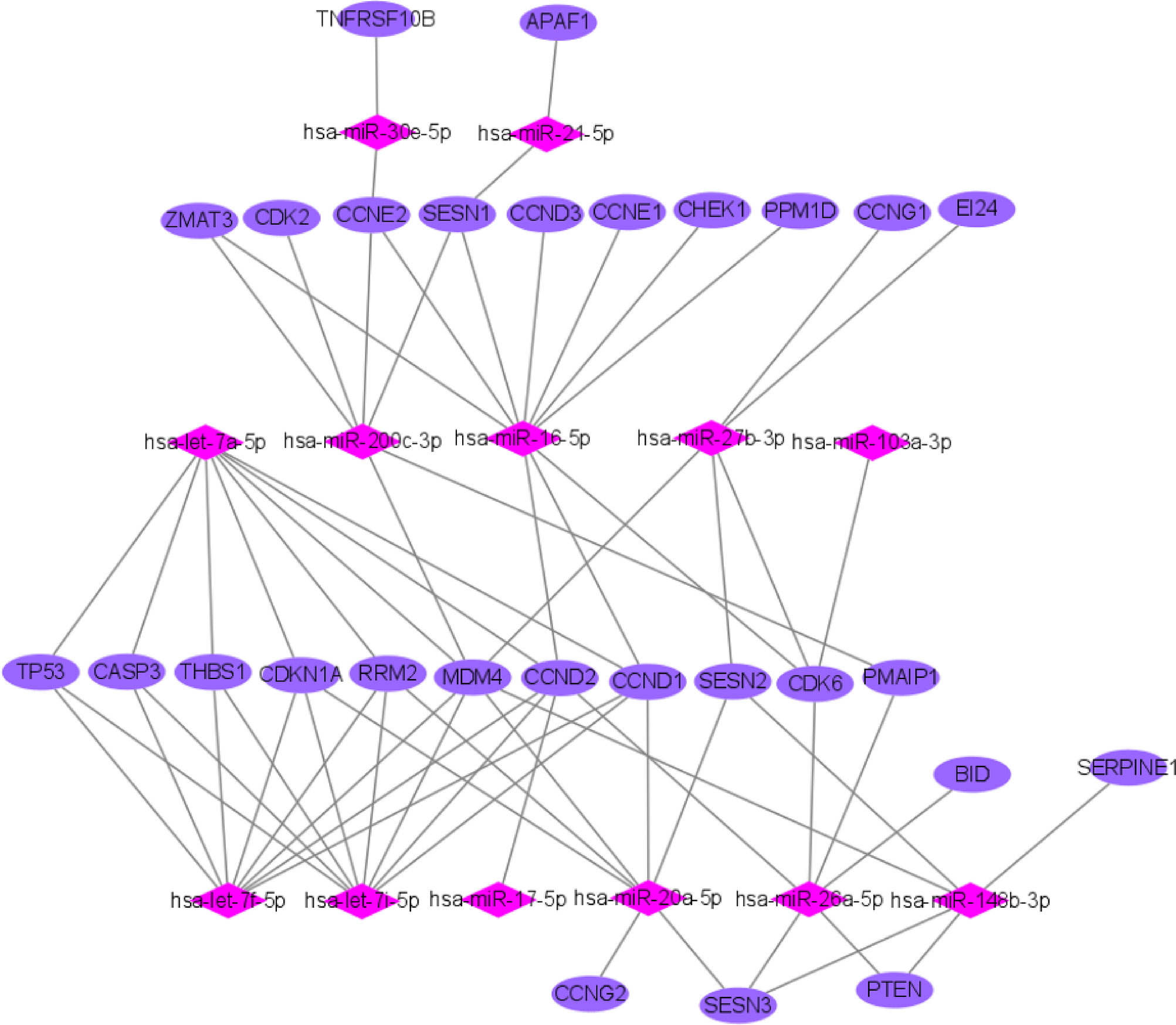
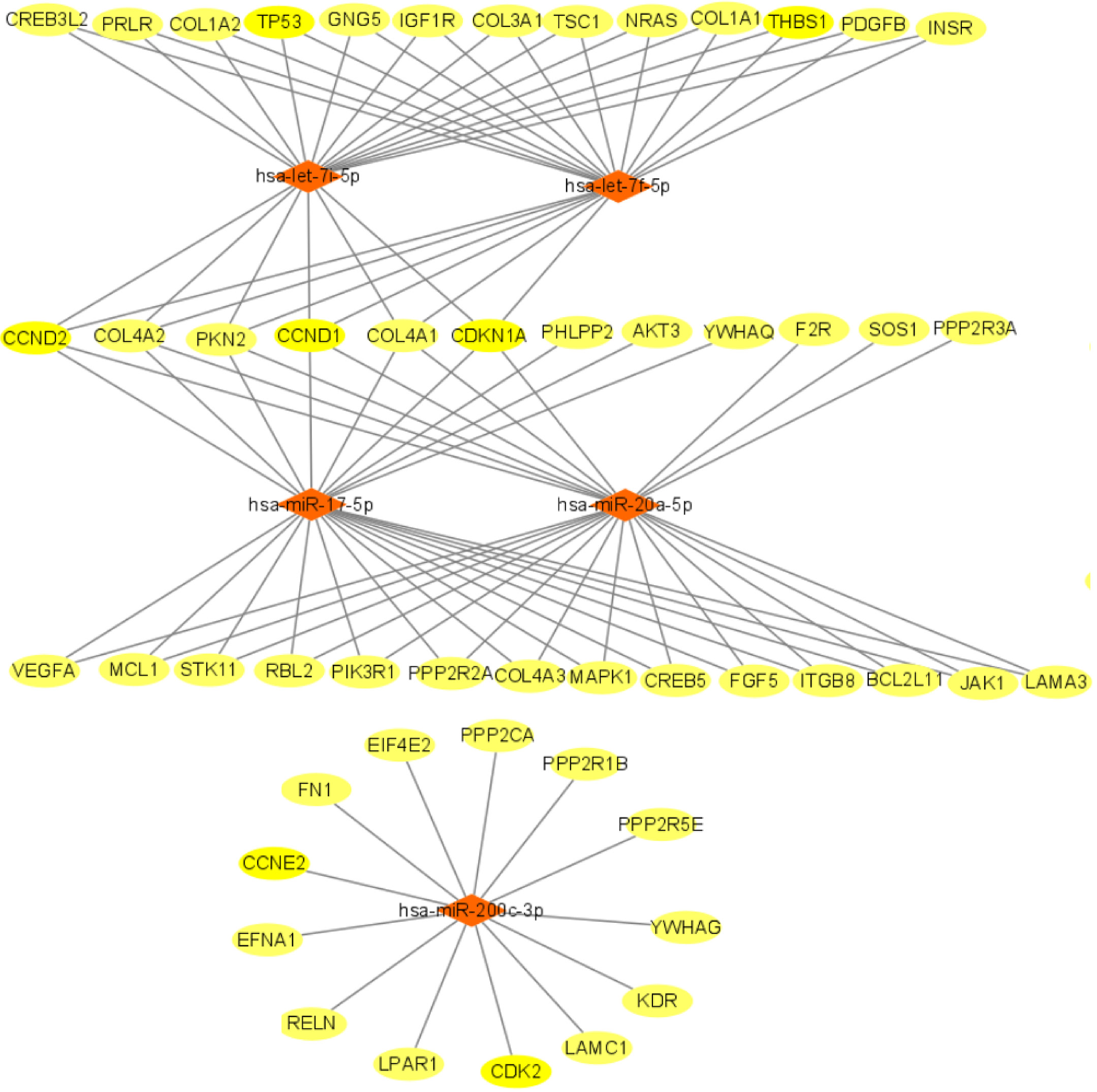
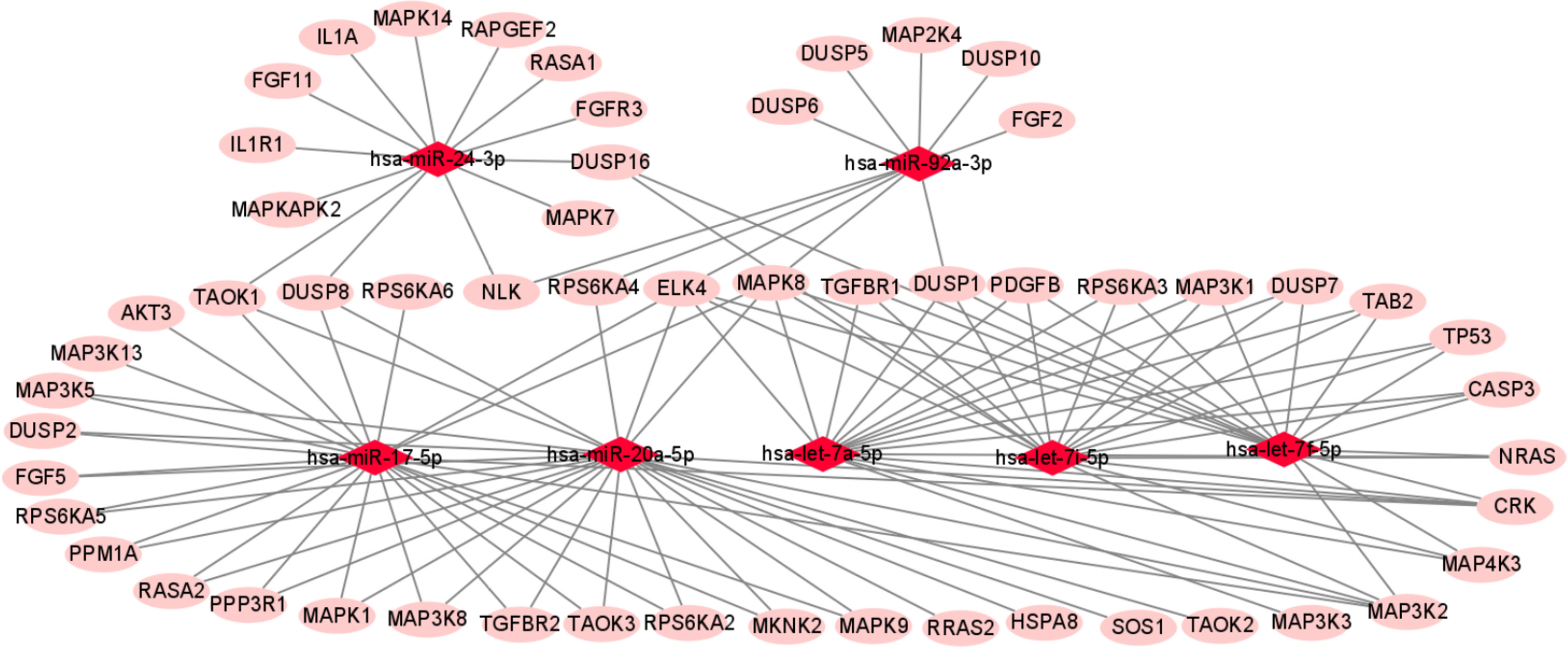
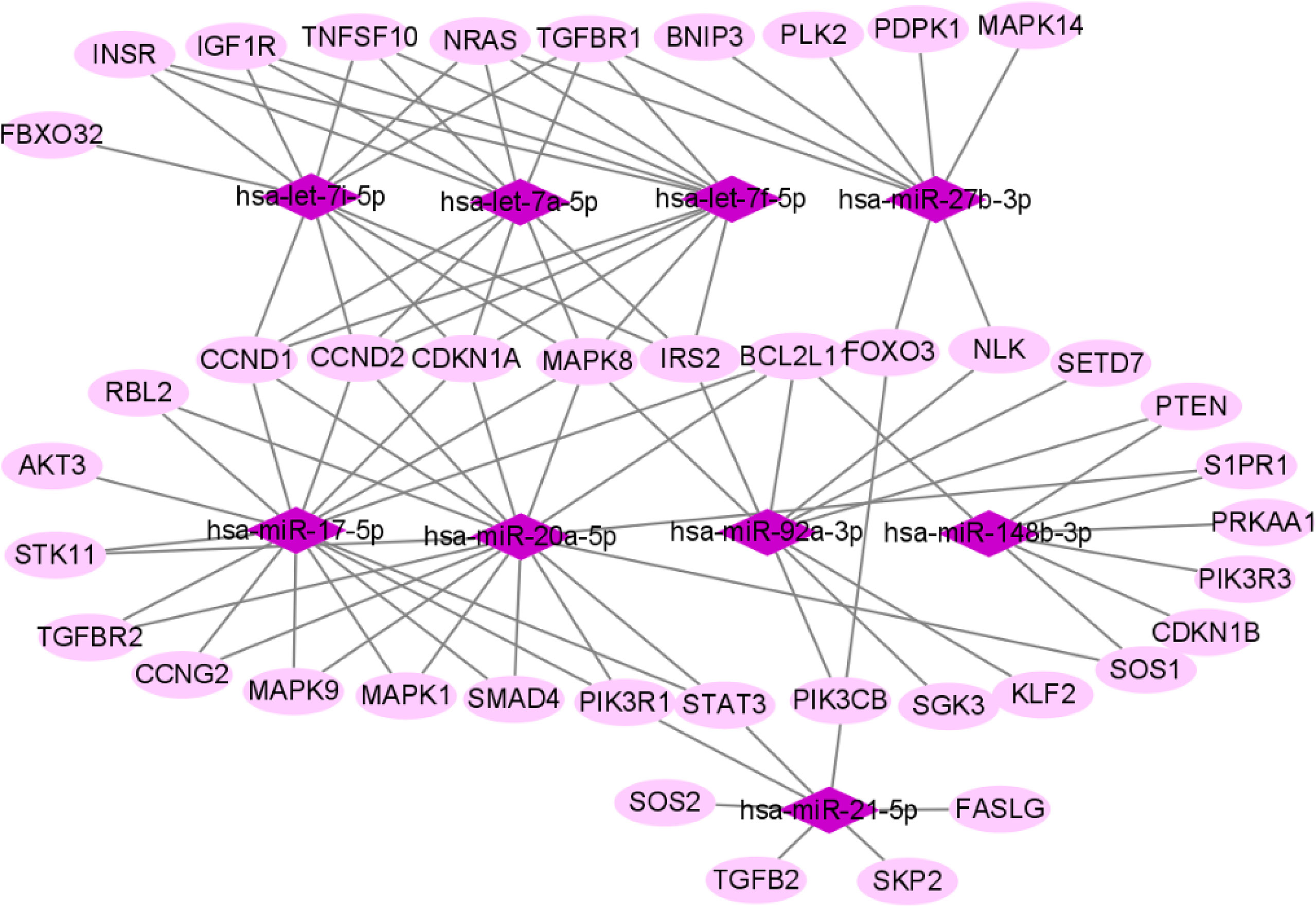
We used miRTarBase and miRDB online software (http://www.targetscan.org/vert_72/ and http://mirdb.org/mirdb/index.html) to predict target genes for the top 20 miRNAs with the highest number of reads in Uniq_reads, enriched the interacting genes in various pathways, and constructed a miRNA-gene interaction network diagram using Cytoscape v. 3.8.0 (https://cytoscape.org/). The cancer, transforming growth factor beta (TGF-β), p53, phosphatidylinositol 3-kinase (PI3K)-protein kinase B (Akt), mitogen-activated protein kinase (MAPK), and forkhead box O (FoxO) signaling pathways play an important role in regulating cancer cell proliferation, apoptosis and migration. The miRNAs that interact with these signaling pathways were selected and network interaction maps were built. The network interaction map for the miRNA-cancer signaling pathway was shown in Figure 3D. The network interaction map for other pathways were presented in Figure 4.
The gene analysis of the interactions of various pathways found that hsa-miR-17-5p and hsa-miR-20a-5p in the cancer, TGF-β, PI3K-Akt, MAPK, and FoxO signaling pathways co-regulated MAPK1, son of sevenless homolog 1 (SOS1) and transforming growth factor beta receptor II (TGFβR2) genes. The hsa-let-7a-5p, hsa-let-7f-5p and hsa-let-7i-5p in the cancer, TGF-β and MAPK pathways targeted and regulated the TGFβR1 gene. The hsa-let-7a-5p, hsa-let-7f-5p and hsa-let-7i-5p in the cancer, MAPK and p53 signaling pathways targeted and regulated the tumor protein 53 (TP53) and caspase 3 (CASP3) genes. The hsa-miR-30e-5p, hsa-miR-92a-3p and hsa-miR-200c-3p in the cancer, p53 and PI3K-Akt signaling pathways targeted and regulated the cyclin E2 (CCNE2) gene. The hsa-miR-92a-3p, hsa-miR-26a-5p and hsa-miR-148b-3p in the cancer, p53 and FoxO signaling pathways targeted and regulated phosphatase and tensin homolog (PTEN) gene. The hsa-miR-200c-3p in the cancer, p53 and PI3K-Akt signaling pathways targeted and regulated the CDK2 gene. The specific miRNA corresponding pathways and target genes are shown in Table 2.
The miRNA-targeted GO and KEGG pathway enrichment analyses
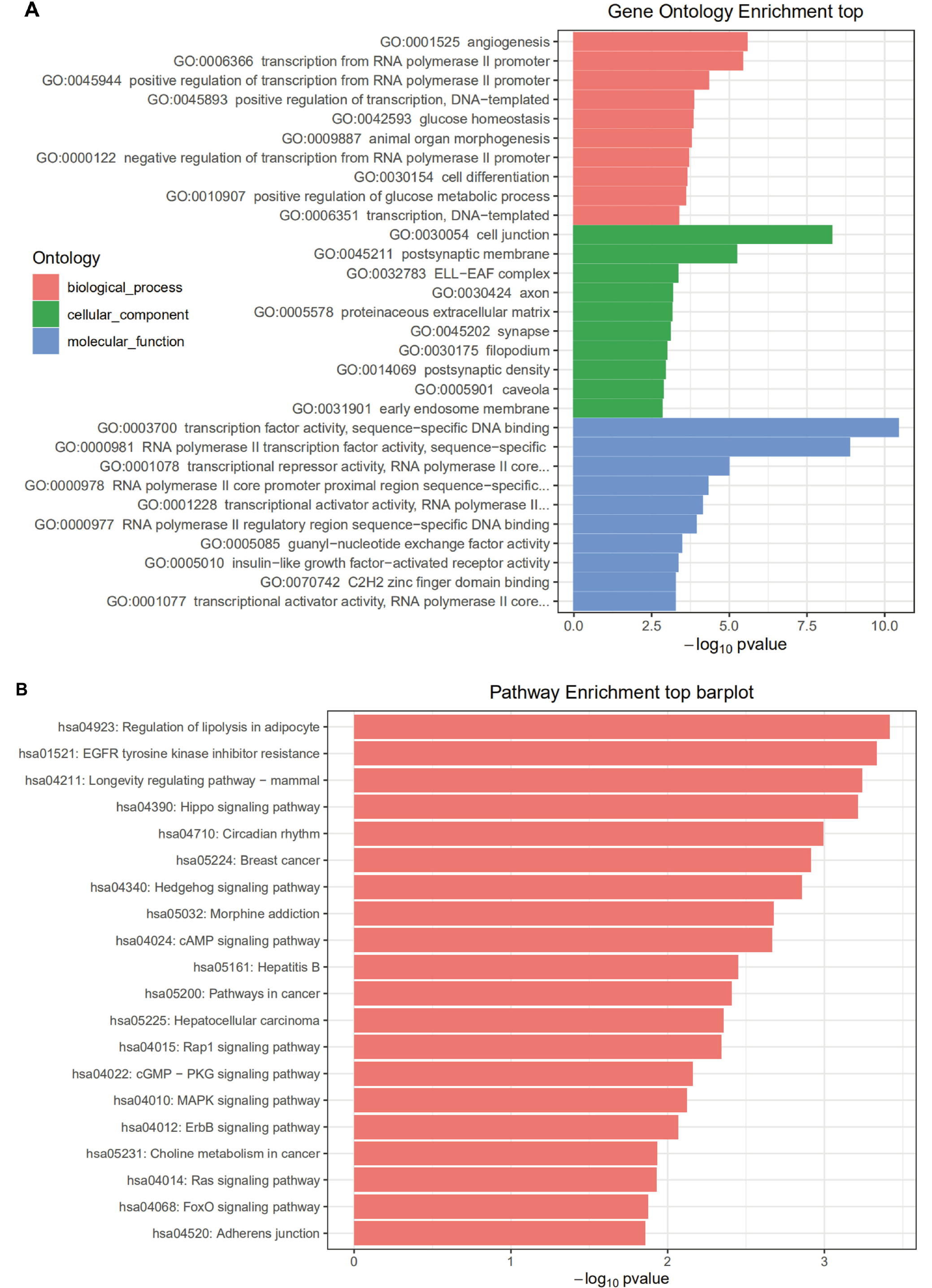
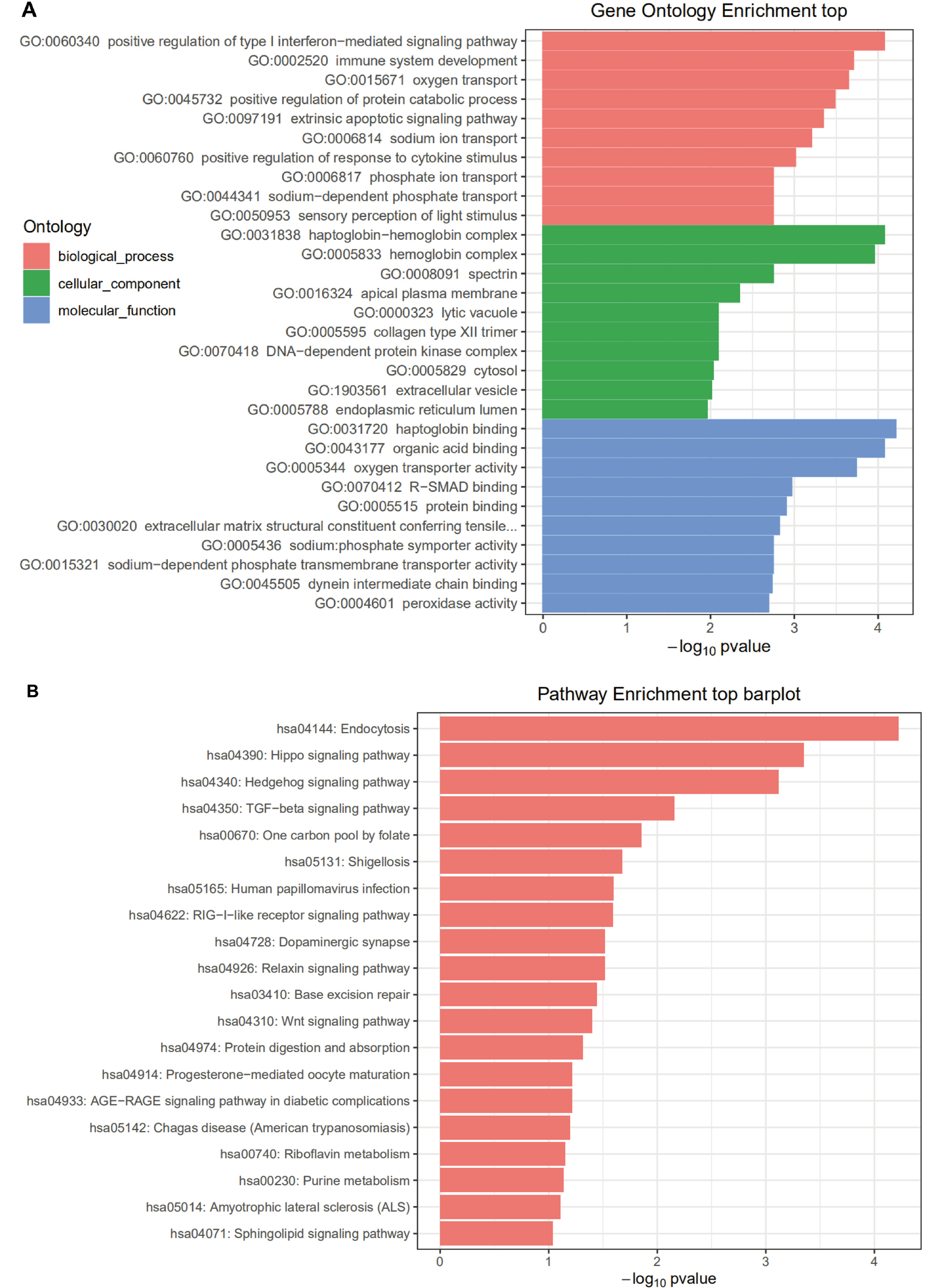
Enrichment analysis was performed on HONE1_M vs. CNE2_M differentially expressed miRNA interacting genes. The enrichment paths of the top 10 counts were displayed in a bar graph (Figure 5). As shown in Figure 5, the GO analysis showed that HONE1_M vs. CNE2_M was mainly enriched in biological process such as negative regulation of transcription from RNA polymerase II promoter (GO:0000122), and positive regulation of transcription from RNA polymerase II promoter (GO:0045944), cellular component such as cytosol (GO:0005829), and molecular function such as protein binding (GO:0005515). In KEGG pathway enrichment analysis, the differentially expressed miRNA interacting genes in HONE1_M vs. CNE2_M were enriched in the cancer-related pathways, such as pathways in cancer (hsa05200), MAPK signaling pathway (hsa04010) and Wnt signaling pathway (hsa04310). The GO and KEGG pathway enrichment analyses for other groups are shown in Figure 6, Figure 7, Figure 8.
Discussion
At present, NPC is mainly treated with radiotherapy, but the treatment effect is not very satisfactory. Most people with NPC will relapse and need to undergo radiotherapy again, which cause a heavy burden to the patient and economy. In radiotherapy, some cancer cells will resist radiation, causing reduction in the apoptosis rate of cancer cells. This resistance is related to the regulation of gene expressions in cell proliferation, apoptosis and migration by miRNA.15, 16, 17 The miRNA enhances the reproductive ability of cancer cells18 and increases the resistance to radiation by downregulating the apoptotic genes of cancer cells, upregulating the expression of proliferation genes or disrupting the cell cycle. There are several cancer cell lines in NPC. In our preliminary experiments, we screened out the X-ray-sensitive CNE2 and the X-ray-insensitive HONE1 cell lines. We performed high-throughput sequencing on these 2 types of irradiated cell lines and explored which miRNAs between the different cell lines regulate related genes to resist X-ray therapy.
We sorted out the data of 4 groups – CNE2_C, CNE2_M, HONE1_C, and HONE1_M – for high-throughput sequencing quality control. After screening by miRNA length and removing duplicate data, we found that the number of reads of CNE2_C and CNE2_M was higher than that of HONE1_C and HONE1_M, and at the same time, we performed boxplot analysis and heat map analysis on the expression levels. The boxplot showed that the median value of CNE2_M was lower than the other 3 groups. There were relatively more low expression miRNAs in CNE2_M, but because there were outliers with relatively high number of reads, the miRNA expressions in CNE2_M were relatively high, and the heat map cluster analysis also showed that the amount of miRNAs with high expression were relatively higher. At the same time, the sequenced RNA species were annotated, and among them, miRNA and rRNA accounted for a relatively larger proportion of the Cleandata reads count.
We conducted a detailed analysis of the top 20 miRNAs in terms of expression, predicted the miRNA family and performed cluster analysis of the miRNA expression levels. The 3 families of mir-21, mir-10 and let-7 had the highest number of miRNA reads, which was consistent with the results of the heat map analysis. In the heat map analysis, the expression level of miRNA-21-5p in the HONE1_M group was higher than that of the CNE2_M and HONE1_C groups. Flow cytometry experiments showed that the apoptosis rate of HONE1_M was lower than that of the CNE2_M group. It is speculated that miRNA-21-5p is related to radiation resistance and inhibition of apoptosis in HONE1_M cells. Studies have found that miR-21 can enhance the resistance of cancer cells to radiotherapy.19 The expression of miR-21 is increased in NPC tissues, downregulated the expression of B-cell lymphoma 2 (bcl2), and inhibited cell migration.20 The miR-21 and miR-205 enhance the resistance of cancer cells to radiotherapy by regulating the PTEN signaling pathway.21, 22, 23 The EBV infection can lead to NPC. The proto-oncogene latent membrane protein (LMP)1 of EBV can upregulate miR-21, negatively regulate the pro-apoptotic factor programmed cell death (PDCD)4 and Fas ligand (Fas-L), and increase the resistance of NPC cells to cisplatin.24 The expressions of let-7i-5p and let-7a-5p of the let-7 family in the 2 groups of HONE1 were higher than in the CNE2 cells. It is speculated that let-7 is related to the resistance of HONE1 to X-rays. The let-7 family is one of the best known miRNA families in cancer biology. It inhibits the proliferation of NPC cells by downregulating the expression of c-Myc.25 The X-rays can enhance the expression of let-7 in NPC cells and inhibit the expression of miR-7, which may increase the radiosensitivity of NPC cells.26
In the heat map analysis, the high expression of hsa-miR-125a-5p in HONE1_M appeared red, and the low expression in CNE2_M appeared green. This differential expression implied that hsa-miR-125a-5p plays an important role in the resistance of cancer cells to radiotherapy. Studies have found that miR-125a-5p can increase p53 protein expression in HNE-1 cells, reduce human epidermal growth factor receptor 2 (Her2) protein expression in HNE-1 and HK-1 cells,27 and promote the proliferation, migration and invasion of HONE1 cells. The expression level of miR-125a-5p in NPC patients was significantly higher than that in healthy controls.28
In the heat map analysis, the expression levels of hsa-miR-26a-5p, hsa-miR-24a-3p and hsa-miR-20a-5p in CNE2_M were high and appeared red, while the expression levels in HONE1_M were low and appeared green. We speculated that these 3 miRNAs are related to the resistance of cancer cells to radiation therapy. The miR-26a has the effect of inhibiting the growth of NPC cells; miR-26a can significantly downregulate the expression of enhancer of zeste homolog 2 (EZH2),29 activate p14 (ARF) and p21 (CIP1), maintain the cell cycle in the G1 phase, enhance the sensitivity of cell to radiation, and inhibit the growth and migration of cancer cells.29, 30 Radiation therapy damages the DNA of NPC cells. Cancer cells can repair part of the damage and inhibit apoptosis. The miR-24 can bind to Jun activation domain-binding protein 1 (Jab1) to inhibit the repair of DNA damage and increase the sensitivity of NPC cells to radiotherapy.31 Some studies have found that the expression of miR-24 is decreased in patients with advanced NPC, which reduces the sensitivity to radiotherapy.31, 32 The miR-20a-5p can regulate the expression of neuronal PAS domain protein (NPAS)2 and Rab27B to reduce the sensitivity of NPC cells to radiotherapy.33, 34 In this study, the expressions of hsa-miR-26a-5p, hsa-miR-24a-3p and hsa-miR-20a-5p in the HONE1_M group were lower than in the CNE2_M group. We speculated that it is likely that the combined action of these 3 miRNAs co-downregulated the sensitivity of cells to X-rays in the HONE1_M group.
We predicted the target genes of the top 20 miRNAs with the highest number of reads in Uniq_reads, and predicted the target genes. The miRNAs that interact with the cancer, TGF-β, p53, PI3K-AKT, MAPK, and FoxO signaling pathways were selected and the network interaction maps were constructed (Figure 3D, Figure 4). The results showed that 9 genes were co-regulated by miRNA in various pathways. These results were also reflected in the GO and KEGG pathway enrichment analyses. Here, we discuss the functions of several genes that are closely related to cancer. The hsa-miR-17-5p and hsa-miR-20a-5p targeted MAPK1, SOS1 and TGFβR2 genes. The MAPK1 is an oncogene, and its expression is upregulated in various cancer tissues. The miR-511 targeting binding with MAPK1 can inhibit the proliferation and invasion of osteosarcoma in nude mice.35 The SOS1 is a Ras-GEF family protein and an oncogene. Knockout of SOS1 protein in mice can inhibit tumor growth and migration.36 In NPC, miR-93 targets TGFβR2, downregulates TGFβR2 expression, and promotes cancer cell proliferation, invasion and migration.37 The hsa-let-7a-5p, hsa-let-7f-5p and hsa-let-7i-5p target and regulate the expression of tumor suppressor genes TP53 and CASP3 in the MAPK and p53 signaling pathways. Studies have found that tripartite motif-containing protein 21 (TRIM21) inhibits the expression of TP53 and protects cancer cells from radiation-induced apoptosis by mediating the ubiquitination and degradation of guanine monophosphate synthase (GMPS).38 The CASP3, also known as caspase 3, is an apoptotic protein, and its mutation can lead to cell carcinogenesis.39 The hsa-miR-92a-3p, hsa-miR-26a-5p and hsa-miR-148b-3p targeted the PTEN gene. The PTEN signaling pathway is related to the resistance of NPC cells to radiotherapy. The inactivation of PTEN activates PI3K-AKT3 signaling pathway and inhibits cell apoptosis.23 The hsa-miR-30e-5p, hsa-miR-92a-3p and hsa-miR-200c-3p targeted the CCNE2 gene. In various cancers, many miRNAs can bind to CCNE2 to inhibit the proliferation of cancer cells. For example, in glioma, miR-370 can downregulate the CCNE2 expression and inhibit tumor growth.40 These genes are directly or indirectly related to cancer proliferation, apoptosis and other physiological processes. They are the key genes for studying miRNA regulation of cancer cell resistance to X-rays.
Limitations of the study
This study mainly investigated the miRNA profiling of HONE1 and CNE2 after X-ray therapy. Further studies may explore the mechanisms involved.
Conclusions
Through an analysis of the miRNA expression and the cluster analysis of the regulatory pathway, 12 miRNAs and 9 genes which play an important role in X-ray radiation resistance were identified. Among those with differential expression between the HONE1 and CNE2 cell lines, which played a regulatory role in multiple pathways, were hsa-miR-20a-5p, hsa-let-7a-5p, hsa-let-7f- 5p, hsa-let-7i-5p, hsa-miR-30e-5p, hsa-miR-148b-3p, and hsa-miR-200c-3p. The corresponding genes were MAPK1, SOS1, TGFBR1, TGFBR2, TP53, CASP3, CCNE2, PTEN, and CDK2. We will conduct more in-depth research on these genes and miRNAs in follow-up studies to provide more experimental evidence for the mechanism of NPC resistance to radiotherapy.