
















Abstract
Drug-resistant epilepsy (DRE) presents a major clinical and economic challenge, particularly in low- and middle-income countries (LMICs), where healthcare resources are limited and treatment gaps remain significant. Although epilepsy surgery remains the most effective intervention for eligible DRE patients, outcomes are variable, with success rates ranging from 30% to 70%. Emerging evidence suggests that genetic biomarkers can inform patient selection, predict surgical outcomes, and guide treatment planning. This review explores the potential of integrating genetic testing into presurgical evaluation protocols in LMICs. It examines the role of specific gene mutations in pharmacoresistance, seizure localization, and structural brain abnormalities, with a focus on improving surgical success rates and reducing unnecessary interventions. Incorporating genetic stratification into clinical decision-making could enhance cost-effectiveness, minimize the burden on healthcare systems, and support the development of personalized treatment pathways. Advancing genetic research and building capacity in precision neurology are essential steps toward improving DRE management in resource-constrained settings.
Key words: drug-resistant epilepsy, surgical outcomes, low- and middle-income countries, genetic testing, patient selection
Introduction
Epilepsy is one of the most common neurological disorders, affecting approx. 50 million individuals worldwide.1, 2 The condition manifests as recurrent unprovoked seizures, which are diagnosed after 2 unprovoked episodes or following a single seizure with a high risk of recurrence.3 The World Health Organization’s Global Action Plan 2022–20314 emphasizes the urgent need for improved epilepsy management, particularly in low- and middle-income countries (LMICs), where approx. 80% of people with epilepsy reside.5
Drug-resistant epilepsy (DRE) is defined by the failure of adequate trials of at least 2 appropriately selected and tolerated antiepileptic drugs to achieve sustained seizure control and presents significant challenges.6 Drug-resistant epilepsy is correlated with increased morbidity, diminished quality of life, and elevated healthcare costs.7 Although epilepsy surgery is the most effective treatment option for selected DRE cases, surgical outcomes vary considerably, with seizure freedom rates ranging from 30% to 70%.8, 9 This variability highlights the need for improved patient selection methods and enhanced understanding of factors influencing surgical success.
Genetic factors are increasingly recognized as key contributors to epilepsy susceptibility, severity, and treatment resistance. Mutations affecting ion channel function, synaptic transmission, cortical development, and neuroinflammation have been implicated in a broad spectrum of epilepsy syndromes.10, 11 These findings have opened the door to the use of genetic data in clinical decision-making, particularly in the context of presurgical evaluation.12, 13 The National Society of Genetic Counselors in the USA recommends genetic testing for all patients with unexplained epilepsy.14 While testing is commonly prioritized for early-onset, syndromic, or developmental epilepsies, its role is expanding in focal DRE as well. For example, recent studies demonstrate that genetic diagnoses can directly influence management and outcomes, leading to treatment modifications in up to 45% of children with severe epilepsy – with around 60% of those receiving gene-directed therapies achieving seizure freedom – and informing clinical decisions in adults, where diagnostic yields range from 23% to 50%.15, 16 Specific findings now inform individualized approaches, such as avoiding sodium channel blockers in SCN1A-related epilepsy or using the ketogenic diet in SLC2A1 cases, while emerging biomarkers like circulating microRNAs and protein profiles help distinguish drug-resistant from drug-sensitive epilepsy.17, 18, 19, 20 Despite growing interest, the incorporation of genetic biomarkers into routine epilepsy care remains limited in many LMICs because of infrastructure, cost, and workforce constraints.
This review summarizes the current evidence on the role of genetic biomarkers in DRE management, focusing on presurgical evaluation and treatment outcomes. Special attention is given to the challenges and opportunities in LMICs, where cost-effective, personalized approaches to epilepsy care are particularly critical. Through synthesis of the current literature, this review provides an overview of the genetic influences on DRE and explores how genetic testing can complement existing diagnostic strategies.
Objectives
The objective of this review is to explore how genetic testing can support presurgical evaluation in DRE, particularly in LMICs. It addresses the clinical impact, economic considerations, and feasibility of applying genetic screening to guide treatment decisions in resource-limited settings.
Materials and methods
A literature search was conducted across PubMed, Scopus, Web of Science, and Google Scholar. No restrictions were placed on publication date. Search terms included “drug-resistant epilepsy,” “genetic testing,” “epilepsy surgery,” “presurgical evaluation,” “clinical utility,” “diagnostic yield,” “cost-effectiveness,” and “low- and middle-income countries.”
The inclusion criteria comprised full-text, peer-reviewed articles in English describing the role of genetic testing in DRE, particularly in the context of presurgical decision-making and treatment planning. Studies involving both pediatric and adult populations were considered. Case reports, cohort studies, reviews, and expert consensus documents were included if they discussed the diagnostic yield, clinical utility, or cost-related implications of genetic testing. Editorials, commentaries, abstract-only records, and studies focusing exclusively on syndromic epilepsies without implications for surgical evaluation were excluded.
Due to the absence of studies conducted specifically in LMICs, this review incorporated findings from high-income countries (HICs), where most genetic testing protocols and outcome data are currently available. These studies were selected for their potential applicability to LMIC settings, especially regarding implementation strategies, diagnostic pathways, and resource prioritization. A systematic review was not feasible because of the limited number of regionally relevant studies and the heterogeneity of study designs and outcome reporting.
Genetic contributors to drug-resistant epilepsy
Genetic variants underlie many cases of severe and focal epilepsy. Ion channel variants, particularly in sodium channels (SCN1A, SCN2A, SCN8A) and potassium channels (KCNQ2/3), represent the most extensively studied genetic determinants of treatment response. De novo or inherited mutations in these genes cause early-onset epileptic encephalopathies and Dravet syndrome, which demonstrate inherent pharmacoresistance.21 Recent genome-wide association studies have revealed that the SCN1A rs2298771 polymorphism significantly increases the risk of antiepileptic drugs (AED) resistance, particularly in South Asian populations.22
Synaptic genes (STXBP1, PCDH19, CDKL5) and metabolic genes (SLC2A1, ALDH7A1) also drive DRE through developmental encephalopathies or neurometabolic syndromes.23, 24, 25, 26, 27 Drug transporter genetics have emerged as critical determinants of treatment response, with the ABCB1 gene encoding P-glycoprotein showing polymorphisms (particularly C3435T) that significantly affect carbamazepine and lamotrigine transport across the blood–brain barrier (BBB).28, 29, 30 Additionally, cytochrome P450 variants (CYP2C9, CYP2C19) influence AED metabolism and contribute to treatment resistance through altered drug clearance.17
Mutations in the mTOR pathway genes (TSC1/2, DEPDC5, NPRL2/3, PTEN) are linked to focal cortical dysplasia and tuberous sclerosis, often yielding localized lesions amenable to resection.12, 31 For example, DEPDC5 mutations frequently co-occur with radiologically occult focal cortical dysplasia and predict the presence of surgically treatable malformations. In contrast, germline mutations in channelopathy genes (SCN1A, CNTNAP2, STXBP1) typically indicate diffuse network pathology and poor surgical outcomes.13 Collectively, pathogenic variants in these genes can drive pharmacoresistance and inform the identification of epileptogenic zones.
Recent advances have increasingly highlighted the critical role of epigenetic modifications in the development and progression of DRE.29, 32 Specifically, DNA methylation patterns exhibit disease-specific alterations in hippocampal tissue obtained from patients with drug-resistant temporal lobe epilepsy.33 These epigenetic changes influence the expression of genes encoding ion channels and neurotransmitter receptors, thereby contributing to the dysregulation of neuronal excitability and synaptic transmission characteristic of refractory seizures. In parallel, microRNA profiling has uncovered a set of circulating small non-coding RNAs, including miR-134-5p, miR-122-5p, and miR-132-3p, that reliably differentiate drug-resistant from drug-sensitive epilepsy patients.19 These microRNAs are implicated in key neurobiological processes such as synaptic plasticity, neuroinflammation, and neuronal survival, all of which play pivotal roles in epileptogenesis and pharmacoresistance. The identification of these epigenetic markers offers promising avenues for developing minimally invasive diagnostic tools and novel therapeutic targets tailored to overcome drug resistance in epilepsy.
The growth of exome/genome sequencing has expanded the list of epilepsy genes; however, practical testing often focuses on established candidates. The ClinGen Epilepsy Gene Curation panel classifies approx. 30 genes as definitive causes of epilepsy (SCN1A, KCNQ2, TSC1/2, DEPDC5, SCN2A, SLC2A1), with many others supported by moderate evidence.34 In pediatric DRE cohorts, diagnostic yields of targeted epilepsy gene panels range from around 12% to 29%, rising to 39–50% in infants with developmental encephalopathies. Whole-exome sequencing (WES) achieves even higher yields – up to 50% in early-onset, syndromic cases.35 Notably, yields are higher when testing is performed early; 1 study found that testing at epilepsy onset halved the cost of the diagnostic workup by avoiding redundant tests.36 Thus, for selected patients (early-onset, multifocal features, family history), genetic testing is highly informative (Table 1).
Genetic testing in the presurgical evaluation of DRE
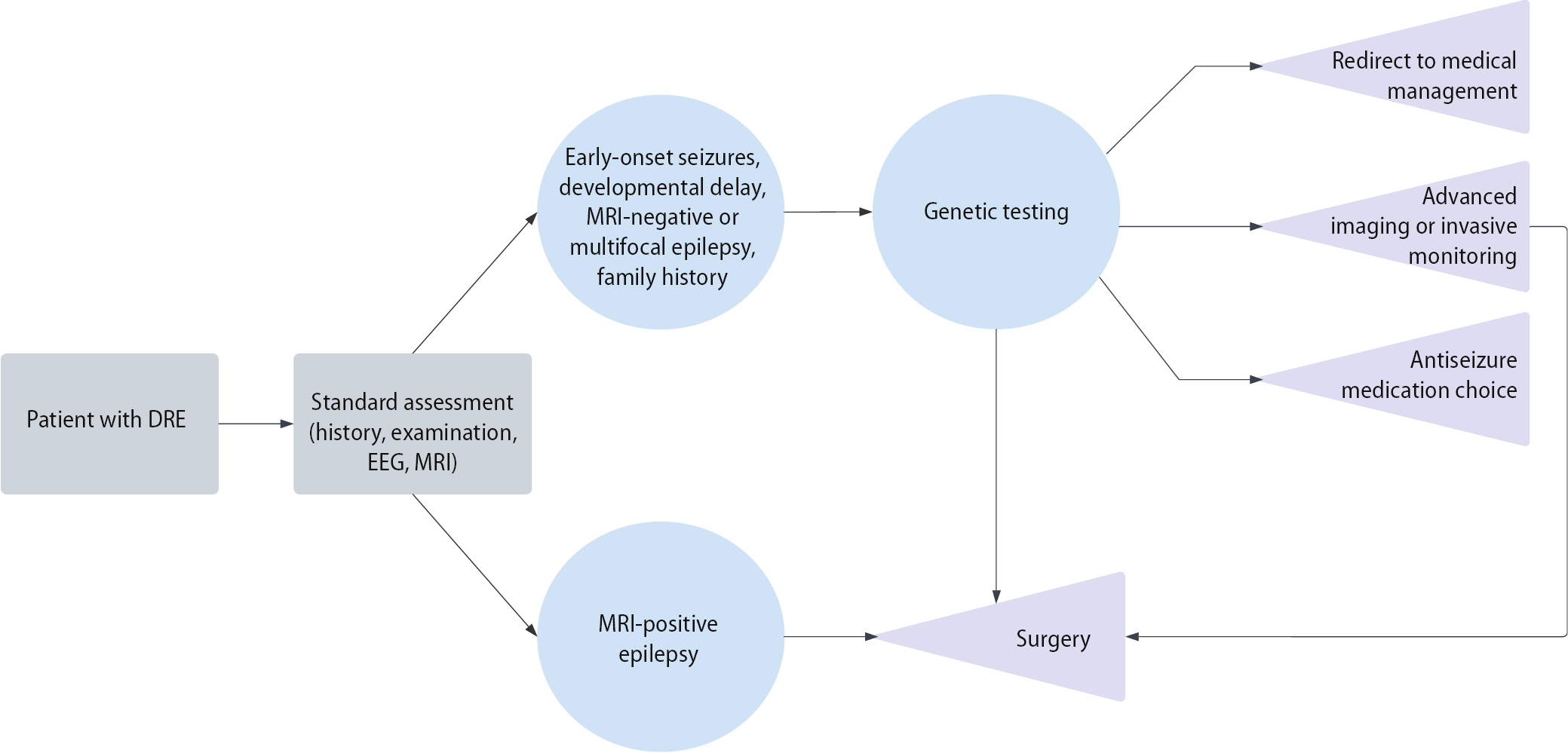
Presurgical evaluation in DRE traditionally relies on clinical assessment, neuroimaging, and electroencephalography (EEG) to pinpoint the epileptogenic zone. However, even with these tools, surgical outcomes remain suboptimal in many patients, with seizures often persisting after surgery.21, 64, 65 The addition of genetic data to this approach is an emerging paradigm. A novel approach to improve these outcomes is the integration of genetic testing into the presurgical workup (Figure 1). Growing expert consensus and recent studies suggest that genetic testing should be considered in patients with focal epilepsy of unknown etiology – particularly those with early-onset seizures, developmental delay, or magnetic resonance imaging (MRI)-negative findings – where genetic etiologies may influence both surgical decision-making and outcomes.66, 67 Large studies underscore this shift: in a 5-year single-center cohort of 125 pediatric DRE surgical referrals, 69% underwent genetic testing, and 21% had a pathogenic variant identified.68 The identified genes included NPRL3, TSC2, KCNH1, CHRNA4, SPTAN1, DEPDC5, SCN2A, ARX, SCN1A, DLG4, and STXBP1. Notably, in 3 cases, the genetic diagnosis directly altered management: those patients did not proceed to surgery because the findings indicated that a surgical cure was unlikely. The authors emphasized that obtaining a molecular diagnosis can significantly impact treatment decisions and suggested that genetic testing should be integrated into the routine assessment process for pediatric patients with DRE during presurgical evaluation.
Other centers have reported similar findings. In 1 cohort, routine WES and microarray analysis of 49 children undergoing epilepsy surgery identified variants in 45% (21 of 49) of the children.13 TSC1/2, DEPDC5, MECP2, and KCNQ2, among others, were identified by both exome and chromosomal microarray analyses. Although the genetic findings did not change whether surgery was performed, they guided postoperative management: patients with mutations were counseled more conservatively, retaining anti-seizure medications for longer. Notably, among the few patients with poor surgical outcomes (International League Against Epilepsy (ILAE) class IV–V), a majority had underlying genetic variants or multifocal syndromes. This suggests that detecting a genetic cause can inform prognosis, guide patient counseling, and aid postoperative planning, even if it does not alter the immediate surgical plan.
Further evidence comes from smaller series. In a focused study of 9 children evaluated for focal epilepsy surgery, targeted epilepsy gene testing led to a major change in management in most cases: 7 of the 9 patients had their surgical workups halted entirely. All 7 had early-onset seizures with developmental delay, and each harbored a pathogenic germline variant in a known epilepsy gene.67 The investigators recommend that genetic assessment should be incorporated as a mandatory component of presurgical evaluation, particularly in patients with negative MRI findings or ambiguous lesions. This recommendation aligns with the perspective that genetic diagnoses may contraindicate invasive monitoring or resection procedures, as these conditions typically represent diffuse neurological disease not amenable to focal surgical cure.69
Genetic stratification can improve presurgical decision-making in several ways. First, it complements imaging and EEG by revealing the underlying mechanism of epilepsy. For example, discovering an mTOR pathway mutation (DEPDC5, TSC1/2) in an MRI-negative patient may prompt the use of invasive EEG (stereo-EEG) to detect subtle focal cortical dysplasia.31 Conversely, identifying a “generalized” genetic epilepsy (e.g., SCN1A, STXBP1) suggests poor localization, leading clinicians to consider medical or neuromodulation therapies instead of surgery.69 Second, genetic findings can directly affect pharmacological management. A well-known example is that SCN1A mutations contraindicate sodium-channel blockers,37 whereas SCN2A/SCN8A mutation (when loss-of-function) may benefit from them.42, 43 Similarly, detecting SLC2A1 (GLUT1 deficiency) guides dietary therapy (ketogenic diet),57 and ALDH7A1 identifies patients treatable with pyridoxine.61 Awareness of such gene-specific therapies can improve seizure control before and after surgery. Finally, knowledge of genetic drug transport or metabolism variants (ABCB1 transporter polymorphisms) may explain resistance and indicate alternative drug choices.70
Mounting clinical evidence reinforces the value of these approaches. Multiple studies have found that obtaining a genetic diagnosis leads to changes in medical management in nearly half of DRE patients and can improve seizure outcomes in a substantial proportion of cases.71 Moreover, certain genetic findings correlate with surgical prognosis: e.g., genetic disorders that cause localized structural abnormalities (such as mTOR-pathway focal cortical dysplasias or mesial temporal sclerosis) are associated with more favorable post-surgical outcomes, whereas patients with genetic generalized epilepsies tend to have poorer seizure control after resection. Taken together, this evidence strongly supports the routine incorporation of genetic testing into presurgical evaluation for DRE – particularly in pediatric patients and those without evident lesions on MRI – to ensure that each patient’s treatment plan is informed by the underlying cause of their epilepsy.
Integrating genetic testing into clinical pathways
Given the benefits genetic information can provide, many epilepsy centers are now integrating genetic testing into their standard clinical pathways for DRE evaluations. In current practice – especially for pediatric DRE cases – a streamlined presurgical workflow has emerged that incorporates genetic testing early in the diagnostic process.36 Following standard clinical, neuroimaging, and electrophysiological assessments, genetic screening is increasingly utilized early in the diagnostic process for individuals meeting high-priority criteria, such as seizure onset before the age of 2, positive family history, failure of 2 or more anti-seizure medications, or normal MRI findings. In many centers, targeted epilepsy gene panels or WES are ordered concurrently with traditional diagnostic evaluations.68, 72 Pathogenic variants identified using these methods may redirect clinical management by precluding invasive monitoring or surgery in cases of diffuse genetic epilepsy or by supporting surgical candidacy in patients with focal lesions. Even when the results are negative or of uncertain significance, they may guide the consideration of additional testing modalities, including assessments for copy number variants or mosaicism. Genetic findings are interpreted in conjunction with neuroimaging and EEG data within multidisciplinary epilepsy teams, which increasingly include expertise in clinical genetics to support integrated decision-making.73
Early experiences with this integrated approach have shown clear advantages. Recent case series highlight that performing genetic testing sooner rather than later in the evaluation can streamline care and reduce unnecessary procedures. For example, 1 study found that when genetic testing was conducted within the 1st year of a child’s epilepsy (early in the disease course), patients required significantly fewer metabolic tests and invasive investigations to reach a diagnosis, compared to those who were tested only after multiple years of DRE.36 In that 5-year series, the overall diagnostic yield of genetic testing was about 12% (28 of 226 patients received a genetic diagnosis). Notably, the subgroup of patients who had early genetic testing accounted for 8 of those diagnoses and avoided numerous additional tests, resulting in lower overall healthcare costs. This suggests that even with a moderate diagnostic yield, early genetic testing can act as a cost-effective triage tool – streamlining the workup by ruling in or out certain etiologies and thereby preventing more costly or invasive diagnostic procedures. As the cost of sequencing continues to fall (WES can now often be performed for under $1,000) and bioinformatics pipelines improve, incorporating genetics early has become increasingly feasible in routine practice.74 Furthermore, economic models from other areas of medicine have shown that first-tier genomic testing can be cost-effective under standard willingness-to-pay thresholds, supporting its use as an upfront diagnostic strategy in appropriate cases.75
In resource-limited environments, the cost-effectiveness of genetic screening must account for local factors. The upfront cost of a gene panel or WES may appear high, but it must be weighed against the costs of failed surgeries, prolonged hospital evaluations, and serial medication trials. For example, epilepsy surgery costs in LMICs range from $500 to $8,000 depending on the country, yet these figures often reflect direct costs without accounting for presurgical evaluations, hospital stays, or postoperative care. Additionally, many clinicians in LMICs are hesitant to refer patients for epilepsy surgery due to limited training and uncertainty about outcomes, which means resources for surgical evaluation may already be underutilized.76 A genetic diagnosis that prevents an unsuccessful surgery can save much more than the test itself. One health economic analyses in pediatric epilepsy (largely from high-income settings) indicated that identifying genetic causes early reduces the diagnostic odyssey and improves long-term outcomes, yielding overall cost.77 Such conclusions likely hold in LMICs, where inefficiencies carry even greater relative burdens.
Overall, the inclusion of genetic testing in standard clinical pathways for DRE allows clinicians to make more informed decisions earlier. This leads to more efficient use of diagnostic resources, prevents patients from undergoing unnecessary invasive procedures or ineffective surgeries, and ensures that each patient’s care plan (medical or surgical) is optimized based on genetic findings.
Challenges and opportunities in LMIC implementation
Despite the promising benefits of genetic testing, implementing these advances in LMICs (Figure 2) presents unique challenges. Healthcare systems in LMICs often face resource constraints that can hinder the adoption of routine genetic testing in epilepsy care. Infrastructure limitations are a major hurdle: advanced neuroimaging (high-resolution MRI) and long-term EEG monitoring are scarce in many regions, and local molecular genetics laboratories are often lacking. A recent global survey of 1,568 providers across 127 countries illustrates the extent of the gap. It found that advanced genetic testing (such as WES or whole-genome sequencing (WGS)) was available to 90.4% of epilepsy care providers in HICs, but only 16.3% of providers in LMICs had access to these technologies.78 In Africa, only 12% reported access to gene panels, whereas in Latin America, the figure was 26% (compared to 63% in Europe). Even where tests exist, most LMICs lack public funding; 37.5% of providers in low-income settings reported that patients must self-fund genetic tests, compared to only 8.6% in high-income settings.78 The high cost of sequencing and limited laboratory infrastructure are clearly key drivers of these disparities.
Another challenge is that genetic data interpretation can be more difficult in LMIC populations. Global reference databases for genetic variants are heavily skewed toward individuals of European ancestry, which means that variants found in patients from other ethnic backgrounds (common in Africa, Asia, and Latin America) are often classified as “variants of uncertain significance” due to a lack of comparative data.79 This uncertainty can limit the clinical actionability of genetic results in non-European populations. Additionally, awareness and training in epilepsy genetics are limited among healthcare providers in many LMICs. Clinicians may not be familiar with when or how to order genetic tests, and there may be cultural or social stigma around genetic disorders that makes families hesitant to pursue testing.80, 81, 82 These factors all contribute to a slower uptake of genetic testing in routine practice.
Despite these barriers, there are still promising developments. International collaborations (ILAE task forces and non-governmental organizations (NGOs)) have established centers of excellence for epilepsy genetics in Africa, Asia, and Latin America (https://www.ilae.org). Telemedicine and cloud-based analysis can compensate for shortages in local expertise.83 Some countries are piloting targeted epilepsy panels using portable sequencers or in partnerships with foreign laboratories.84 Moreover, LMICs can adopt a phased, high-impact approach and prioritize genetic testing for high-yield cases. For example, testing could be reserved for early-onset DRE, patients with a suggestive family history, and cases in which surgery is being considered. In such targeted cohorts, diagnostic yields of genetic testing are often in the 20–40% range, which makes testing cost-effective given the potential to change management. For example, a recent study in Nigeria performed exome sequencing on 22 children with unexplained DRE and achieved a 27% diagnostic rate.85 These 6 diagnoses (genes BPTF, NAA15, SCN1A, TUBA1A, CACNA1A) not only explain epilepsy but also open potential therapeutic avenues (e.g., channel blockers for CACNA1A-related seizures). This kind of focused testing demonstrates that even in LMIC settings, genetic diagnostics can yield clinically meaningful results when applied to high-yield scenarios.
The economic rationale for genetics in LMICs is compelling. Identifying a mutation that predicts surgical failure prevents unnecessary expenditure on invasive evaluations and surgeries for that patient, thereby conserving limited resources. Conversely, identifying resectable genetic epilepsy can help prioritize patients requiring limited surgery slots. As sequencing becomes increasingly accessible, LMICs will benefit from precision methodologies. Key requirements for success in LMICs include capacity-building (training neurologists and geneticists, and educating general clinicians about genetics), securing funding support (through government programs or international donors) to subsidize testing costs, and adapting clinical guidelines to local realities (e.g., creating simplified genetic testing protocols that can be followed in low-resource hospitals). Notably, the WHO’s Global Action Plan on epilepsy (2022–2031) explicitly calls for timely diagnosis and care across all regions,4 implying that access to genetic tools should be part of this global strategy.
Future directions
Looking ahead, the role of genetic testing in managing DRE is set to expand, and several important directions for future development are evident. One critical need is for large multicenter studies that can validate specific genetic biomarkers as predictors of surgical outcomes and inform evidence-based guidelines. Thus far, most studies linking genetics to epilepsy surgery outcomes have been relatively small, focused primarily on pediatric cases, and often single-center. To generalize these findings, future research should include more diverse patient populations (including adults and various ethnic groups) and incorporate data from LMICs, where genetic contributions to epilepsy might differ. These larger studies would enable the development of standardized screening algorithms – tools that help clinicians decide which DRE patients should undergo genetic testing and how to interpret the results in the context of surgical decision-making. Additionally, further cost-effectiveness analyses are needed, especially those tailored to low-resource healthcare systems, to guide policymakers on how to implement genetic testing in a sustainable way.
Emerging technologies will likely accelerate progress in this field. Portable and point-of-care genetic sequencing devices are being developed, which could allow clinics (even in remote areas) to perform genetic tests without relying on distant laboratories. Similarly, advances in bioinformatics and artificial intelligence (AI)-driven variant interpretation are expected to make it faster and easier to distinguish pathogenic mutations from benign variants, addressing one of the current bottlenecks in genomic medicine. As these tools become more affordable and user-friendly, they will help bring genetic diagnostics into mainstream clinical use. At the same time, it will be important to address ethical, cultural, and privacy considerations surrounding genetic testing. Engaging communities to improve understanding of epilepsy genetics, tackling stigma associated with genetic conditions, ensuring informed consent, and establishing clear guidelines for data privacy and sharing are all essential steps as broader implementation of genomic medicine in epilepsy progresses.
While we anticipate these future developments, there are steps that epilepsy programs can take now to begin harnessing genetics. We recommend that multidisciplinary epilepsy centers develop and adopt protocols for selective genetic screening as part of the DRE evaluation. One model is reflexive testing: for instance, automatically sending a blood sample for WES as soon as a patient is diagnosed with DRE (after failing 2 appropriate medications), rather than waiting for multiple failed treatments or inconclusive tests. Another approach is to use targeted gene panels for specific clinical scenarios (e.g., a panel of known infantile epilepsy genes for a child with seizures starting in the 1st year of life).
Moreover, increasing collaboration between hospitals in LMICs and international research consortia can provide interim solutions. Initiatives such as H3Africa (https://h3africa.org) and Central Asian & Transcaucasian Genomics (https://www.cat-genomics.com) are already connecting clinicians in developing regions with resources and expertise in genomics. By partnering with such consortia, a center in a low-resource setting can access genetic sequencing and interpretation services for its patients, even as it works on building local capacity. Importantly, whenever a genetic diagnosis is made, it must be accompanied by appropriate genetic counseling and integrated clinical follow-up. The ultimate goal is not just to identify genes for the sake of knowledge, but to translate that knowledge into better patient care – whether it means altering medications, recommending dietary therapy, advising against unnecessary surgery, or screening family members at risk.
Limitations of the study
This review has several limitations. First, the synthesis is based primarily on studies conducted in high-income settings, which may not be generalizable to LMIC contexts due to differences in healthcare infrastructure, genetic ancestry, and treatment availability. The scarcity of large, population-based studies from LMICs limited our ability to draw region-specific conclusions or assess implementation feasibility in these settings. Many of the included reports were case series or single-center studies with small sample sizes, introducing potential selection and publication biases. Cost-effectiveness data were extrapolated from HICs and may not reflect local economic conditions or healthcare priorities in LMICs. Furthermore, the review did not include a systematic assessment of study quality or risk of bias due to the heterogeneity and limited number of eligible studies. The absence of standardized outcome measures and inconsistent reporting of genetic variants also hindered comparisons across studies. Finally, ethical and sociocultural considerations – such as stigma, informed consent, and data sharing – remain underexplored in the existing literature, particularly in LMIC contexts, and merit dedicated investigation in future research.
Conclusions
Genetic biomarker screening holds significant potential to improve the management of DRE, especially when integrated into presurgical evaluation. The evidence synthesized in this review indicates that incorporating genetic testing into standard workups can refine patient selection for epilepsy surgery and guide more personalized treatment strategies. By identifying underlying genetic etiologies, clinicians are better able to predict which patients are likely to benefit from surgical intervention and which may require alternative approaches, thereby avoiding futile surgeries and optimizing the use of limited resources. This approach is particularly impactful in LMICs, where healthcare resources are constrained and the consequences of unsuccessful interventions are considerable. As genetic testing becomes more accessible and cost-effective, its adoption in presurgical protocols across diverse settings could help close the epilepsy treatment gap. Integrating genetics into routine DRE care paves the way for precision medicine approaches that improve patient outcomes, reduce the burden of uncontrolled seizures, and ultimately enhance the quality of life for individuals with DRE worldwide.
Use of AI and AI-assisted technologies
Not applicable.
.jpg)