Abstract
Background. Abdominal aortic aneurysm (AAA) is a cardiovascular condition characterized by the abnormal dilation of the abdominal aorta.
Objectives. A circular RNA (circRNA) microarray was utilized to identify differentially expressed circRNAs in angiotensin II (Ang II)-stimulated AAA mice.
Materials and methods. Male apolipoprotein E-deficient (apoE–/–) mice were randomly assigned to 2 groups and subjected to 28 days of infusion with either Ang II or saline. At the end of the experiment, the mice were euthanized via exsanguination under anesthesia. The periadventitial tissues were carefully removed from the aortic wall to measure the maximal external diameter of the suprarenal aorta, and then stored for further analysis. Samples from both the control and AAA groups were used for circRNA expression profiling. The R package Bioconductor was employed to perform Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. Arraystar’s proprietary miRNA target prediction software, integrating miRanda and TargetScan, was used to predict the circRNA/miRNA interactions. Reverse transcription quantitative polymerase chain reaction (RT-qPCR) was employed to confirm the reliability of the microarray results.
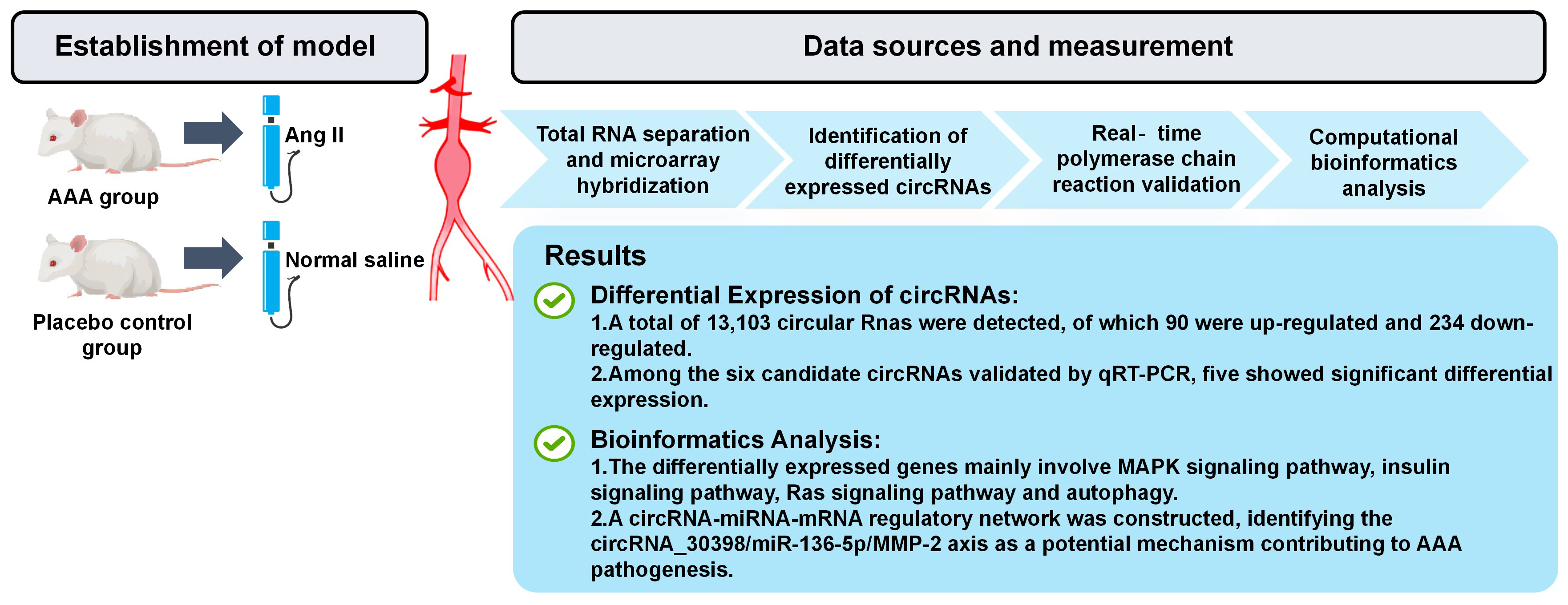
Results. A total of 13,103 circRNAs were detected. Compared to the control group, 90 circRNAs were upregulated and 234 were downregulated in the Ang II-induced AAA group. Gene Ontology analysis indicated that the target genes associated with the differentially expressed circRNAs were involved in a variety of biological processes. The KEGG pathway analysis revealed that the differentially expressed circRNAs influenced several critical pathways, including the MAPK signaling pathway, insulin signaling pathway, Ras signaling pathway, and autophagy. The results of RT-qPCR showed that the expression levels of circRNA_30395, circRNA_30398 and circRNA_012594 were significantly increased in AAA, while circRNA_006097 and circRNA_009932 were notably decreased. The top 5 miRNAs related to each validated circRNA were identified through bioinformatic analysis. Among these differentially expressed circRNAs, miR-136-5p was predicted to be the target gene of circRNA_30398 with high probability.
Conclusions. The differential expression of various circRNAs identified in AAA suggests that the circRNA-miRNA-mRNA axis may serve as a potential molecular regulatory mechanism for AAA.
Key words: gene expression profiling, circular RNA, abdominal aortic aneurysm, angiotensin II, mmu_circRNA_30398
Background
Abdominal aortic aneurysm (AAA) is an asymptomatic and potentially life-threatening cardiovascular disorder, characterized by a localized dilation of the abdominal aortic wall that exceeds 1.5 times its normal diameter or surpasses 3 cm.1 It typically progresses asymptomatically and slowly until rupture occurs. Globally, the prevalence of asymptomatic AAA ranges from 4% to 8%, with rupture occurring at a rate of 4–13 cases per 100,000 individuals annually.2 The condition predominantly affects elderly individuals, particularly those with a history of smoking.3 Once rupture occurs, it can lead to massive internal bleeding, with mortality rates approaching 90%.4
To prevent rupture, surgical repair – either through endovascular aneurysm repair (EVAR) or open repair – is commonly employed. However, both approaches carry considerable perioperative risks and a high rate of postoperative re-intervention.5 From both an economic and clinical standpoint, monitoring and managing small aneurysms to delay or avoid surgical intervention is of greater importance. Based on experiments in rodent models of AAA, successful intervention targets include tissue remodeling, vascular inflammation, lipid metabolism, and blood pressure regulation.5 Although certain clinical studies suggest the possibility of stabilizing growing AAAs through medical intervention, these findings have not been confirmed in larger, controlled trials. Currently, there is no approved pharmacological treatment to slow the progression of small AAAs.6
Circular RNAs (circRNAs) are a class of non-coding RNAs produced through the back-splicing of precursor mRNA exons.7 CircRNAs, with their circular structure and absence of free ends, are more stable than linear RNAs in extracellular plasma, making them well-suited as diagnostic biomarkers.8 Emerging evidence suggests that a subset of circRNAs can act as sponges for microRNAs (miRNAs) due to their miRNA response elements (MREs), thereby preventing miRNAs from binding to their mRNA targets.9, 10 This mechanism is referred to as the competing endogenous RNA (ceRNA) theory.11 Nuclear-resident circRNAs have been shown to influence gene expression at both the transcriptional and splicing levels.12 Recent research has uncovered diverse biological functions of circRNAs in the cardiovascular system, including angiogenesis, vascular smooth muscle cell (VSMC) apoptosis, VSMC proliferation, and endothelial cell migration.13, 14, 15, 16, 17 Given the growing evidence linking circRNAs to cardiovascular regulation, it is plausible that they also play a significant role in the development and progression of AAA.
Notably, the angiotensin II (Ang II)-induced AAA mouse model is the most widely used in vivo system for investigating AAA pathogenesis.18
Objectives
This study aimed to identify differentially expressed circular RNAs (circRNAs) in Ang II-induced AAA mouse models using a circRNA microarray platform. The expression levels of selected circRNAs were further validated by reverse transcription quantitative polymerase chain reaction (RT-qPCR). Additionally, a custom bioinformatics tool, integrating the miRanda and TargetScan algorithms, was used to predict potential circRNA/miRNA interaction networks.
Materials and methods
Study design and participants
Models of mice
Thirty male apoE–/– mice (12 weeks old, C57BL/6 background) were obtained from GemPharmatech Co., Ltd. (Nanjing, China). All animals were housed under specific pathogen-free (SPF) conditions at the Zhejiang Academy of Medical Sciences (Hangzhou, China). Experimental procedures and animal handling were performed in accordance with the Guide for the Care and Use of Laboratory Animals issued by the U.S. National Institutes of Health (NIH Publication number 85–23, revised 1996: National Institutes of Health (NIH), Bethesda, USA), as well as the guidelines approved by the Laboratory Animal Welfare and Ethics Committee of the Zhejiang Academy of Medical Sciences.
Throughout the experiment, the mice were monitored daily for signs of distress, including changes in behavior, body weight and mobility. The mice were randomly assigned to 2 groups. In the AAA group, the mice received a subcutaneous implantation of an Alzet osmotic minipump (model 2004; ALZET Scientific Products, Mountain View, USA) delivering Ang II at a rate of 1,000 ng/kg/min (Sigma Chemical Co., St. Louis, USA) for 28 consecutive days.18 In contrast, mice in the control group received normal saline in the pumps.
Minipump implantation
The procedure for micropump implantation was performed as previously described by Lu et al.19 Prior to implantation, each mouse was weighed, and the midpoint body weight was calculated for each animal to determine the appropriate dosage of Ang II, based on its release rate. The total volume of Ang II required for all mice was subsequently calculated. During the implantation procedure, the mice were anesthetized with isoflurane (Yipin, Shijiazhuang, China), and an incision approx. 1 cm long was made perpendicular to the tail, behind the ear, over the shoulder blade of the foreleg. A subcutaneous pocket was created by inserting the tip of a hemostat toward the tail. The preloaded Alzet osmotic minipump was then carefully inserted into the pocket and advanced fully. The incision was closed with sutures to ensure complete wound closure.
Following surgery, the mice were placed in individual cages with unrestricted access to food and water. Analgesics were provided as needed to manage pain, and the incision site was inspected daily for signs of infection or improper healing. Behavioral assessments were performed regularly to detect any postoperative complications. Mice exhibiting signs of significant distress or health deterioration were evaluated by a veterinarian. Upon confirmation of recovery, the mice were returned to standard housing conditions, and daily welfare checks continued throughout the 28-day Ang II infusion period.
Sample collection
Upon completion of the research, the mice were anesthetized with isoflurane, and blood samples were collected from the right ventricle for subsequent analysis. Following systemic perfusion with cold saline, the aortic tissues were dissected from the ascending aorta to the iliac bifurcation. The gross appearance of the aortic tissues was captured through digital photography after peripheral fat was carefully removed. Immediately thereafter, the cleaned aortic tissues were snap-frozen in liquid nitrogen and stored at –80°C until further analysis.
Data sources and measurement
Total RNA separation and microarray hybridization
Three aortic tissue samples from each of the AAA and sham groups were selected for RNA extraction. Total RNA was isolated using Trizol reagent (Invitrogen Life Technologies, Carlsbad, USA), following the manufacturer’s instructions. RNA concentration and purity were quantified using the NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, USA). Subsequent microarray hybridization and sample preparation were conducted according to the standard protocols provided by Arraystar (Rockville, USA). Briefly, the total RNA samples were treated with RNase R (Epicentre, Inc, Madison, USA) to remove linear RNAs and enrich for circRNAs. The enriched circRNAs were then reverse-transcribed and amplified into fluorescently labeled complementary RNA (cRNA) using the random priming method provided in the Arraystar Super RNA Labeling Kit. The labeled cRNAs were hybridized to the Arraystar Mouse circRNA Array V2 (8 × 15K; Arraystar). After washing, the microarray slides were scanned using the Agilent Scanner G2505C (Agilent Technologies, Santa Clara, USA).
Identification of differentially expressed circRNAs
The acquired array images were processed and analyzed using Agilent Feature Extraction software (v. 11.0.1.1; Agilent Technologies). Quantile normalization and statistical analysis were performed using the limma package in R software (R Foundation for Statistical Computing, Vienna, Austria). A p < 0.05 and a fold change ≥1.5, estimated through a t-test, were used to identify differentially expressed circRNAs between the 2 groups. Discernible circRNA expression patterns among the specimens were visualized via hierarchical clustering.
Real-time polymerase chain reaction validation
To validate the circRNA expression profiles obtained from the microarray analysis, fluorescence-based RT-qPCR was performed using the C1000 system (Applied Biosystems, Waltham, USA). A total of 3 downregulated and 3 upregulated circRNAs were selected for analysis. GAPDH was used as the internal reference gene. The specific primer sequences used for RT-qPCR are listed in Supplementary Table 1. Relative expression levels were calculated using the 2−ΔΔCt method.
Computational bioinformatics analysis
The interactions between differentially expressed circRNAs and their potential target miRNAs were predicted using an in-house miRNA target prediction tool, which integrates both miRanda and TargetScan algorithms. A circRNA/miRNA interaction network involving 5 putative miRNA partners and circRNAs was established based on the predicted miRNA binding sites. To further explore the functional implications of the dysregulated circRNAs, Gene Ontology (GO; https://geneontology.org) enrichment analysis was performed using Bioconductor packages in R, with annotations categorized into biological process, cellular component and molecular function. Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG; https://www.genome.jp/kegg) pathway enrichment analysis was conducted to identify relevant biological pathways associated with the parental genes of the dysregulated circRNAs.
Statistical analyses
The Shapiro–Wilk test was used to assess the normality of data distribution. For comparisons between groups, an unpaired t-test was applied to datasets with a normal distribution. A p < 0.05 was considered statistically significant. All statistical analyses were performed using IBM SPSS v. 23.0 (IBM Corp., Armonk, USA).
Results
Expression profiles of circRNA in mouse tissues of AAA
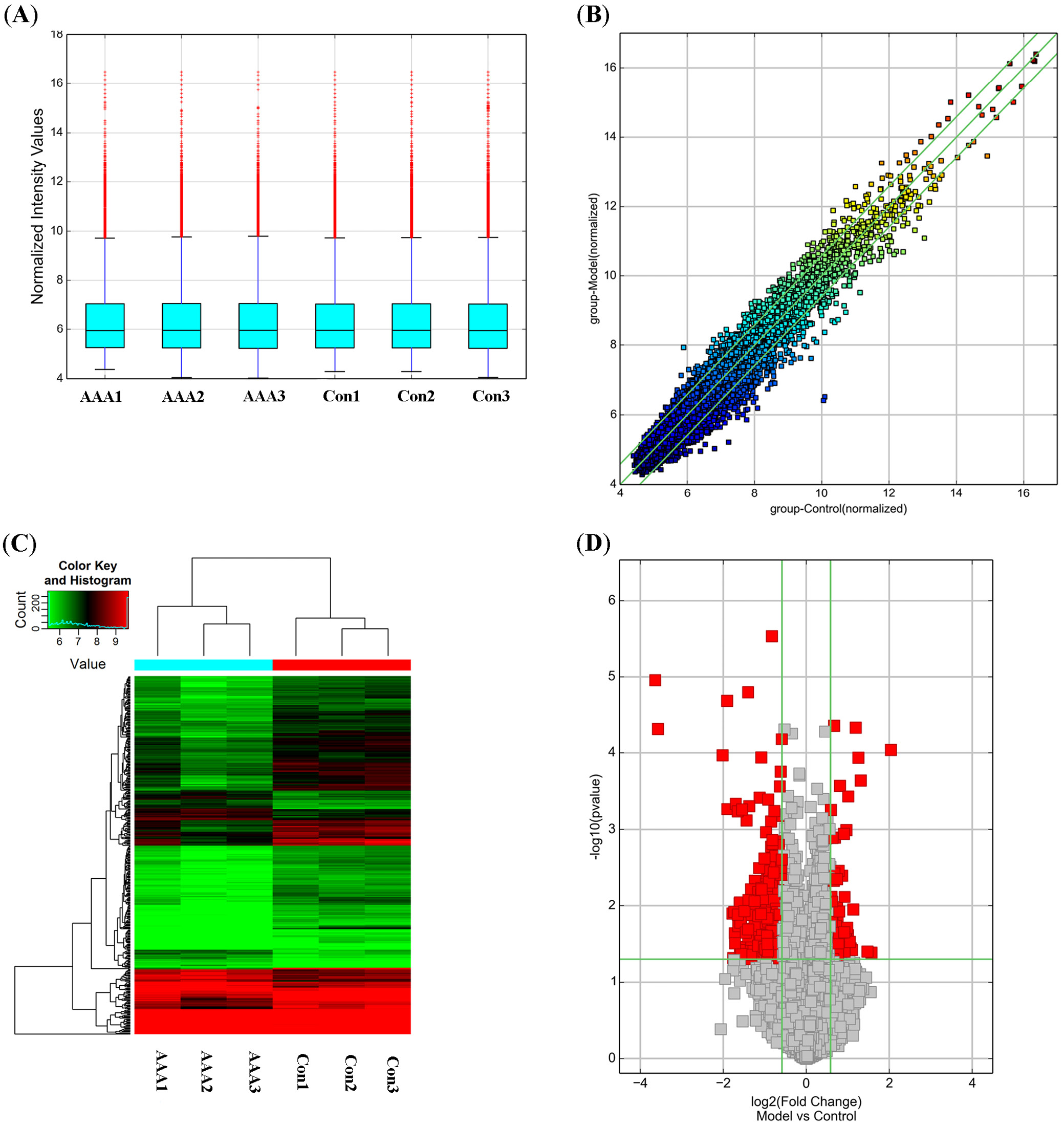
The Ang II-infusion AAA mouse model closely resembles human AAA in several key pathological features, including medial degeneration, atherosclerosis, increased endothelial permeability, thrombus formation, leukocyte and phagocyte extravasation, and matrix sensitivity to proteolysis.20 In this study, AAA was induced via continuous Ang II infusion in apoE–/– mice to examine the circRNA expression profiles (Figure 1). In the AAA group, 7 mice developed typical AAA (with 1 case of rupture-related death), 4 mice exhibited arterial dilation without meeting AAA diagnostic criteria, and 4 mice showed minimal abdominal aorta dilation. In total, 13,103 circRNAs were detected across all samples.
Differentially expressed circRNAs between the Ang II-induced AAA group and the control group were identified using Student’s t-test and fold-change filtering (fold change ≥1.5; p < 0.05), resulting in 90 upregulated and 234 downregulated circRNAs. The volcano plot and hierarchical clustering revealed the circRNA expression profiles between the AAA group and the control group (Figure 1).
Analysis of GO and KEGG pathways in differentially expressed circRNAs
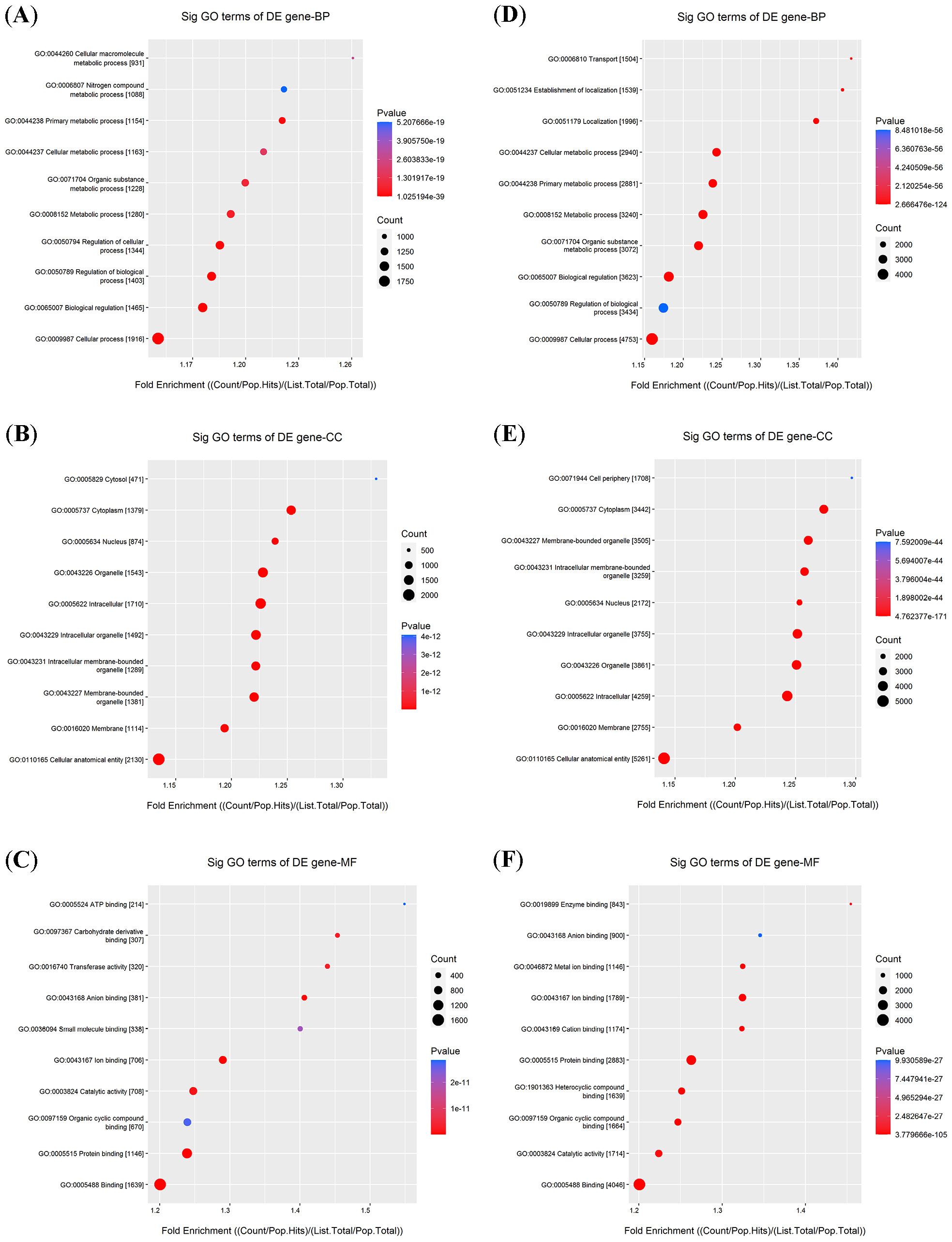
Previous studies have demonstrated that circRNAs can act as miRNA sponges to modulate gene expression. Based on this regulatory mechanism, we performed a bioinformatics analysis using miRanda (https://www.microrna.org)and TargetScan (https://www.targetscan.org) to predict potential miRNA targets of the differentially expressed circRNAs. Subsequently, the predicted target genes were subjected to GO and KEGG pathway enrichment analyses. Gene Ontology enrichment analysis of the upregulated target genes revealed significant enrichment in several processes, including cellular macromolecule metabolism and nitrogen compound metabolism (Figure 2A). Regarding cellular components, these genes were primarily associated with intracellular membrane-bounded organelles and the nucleus (Figure 2B). The most enriched molecular functions were carbohydrate derivative binding and ATP binding (Figure 2C).
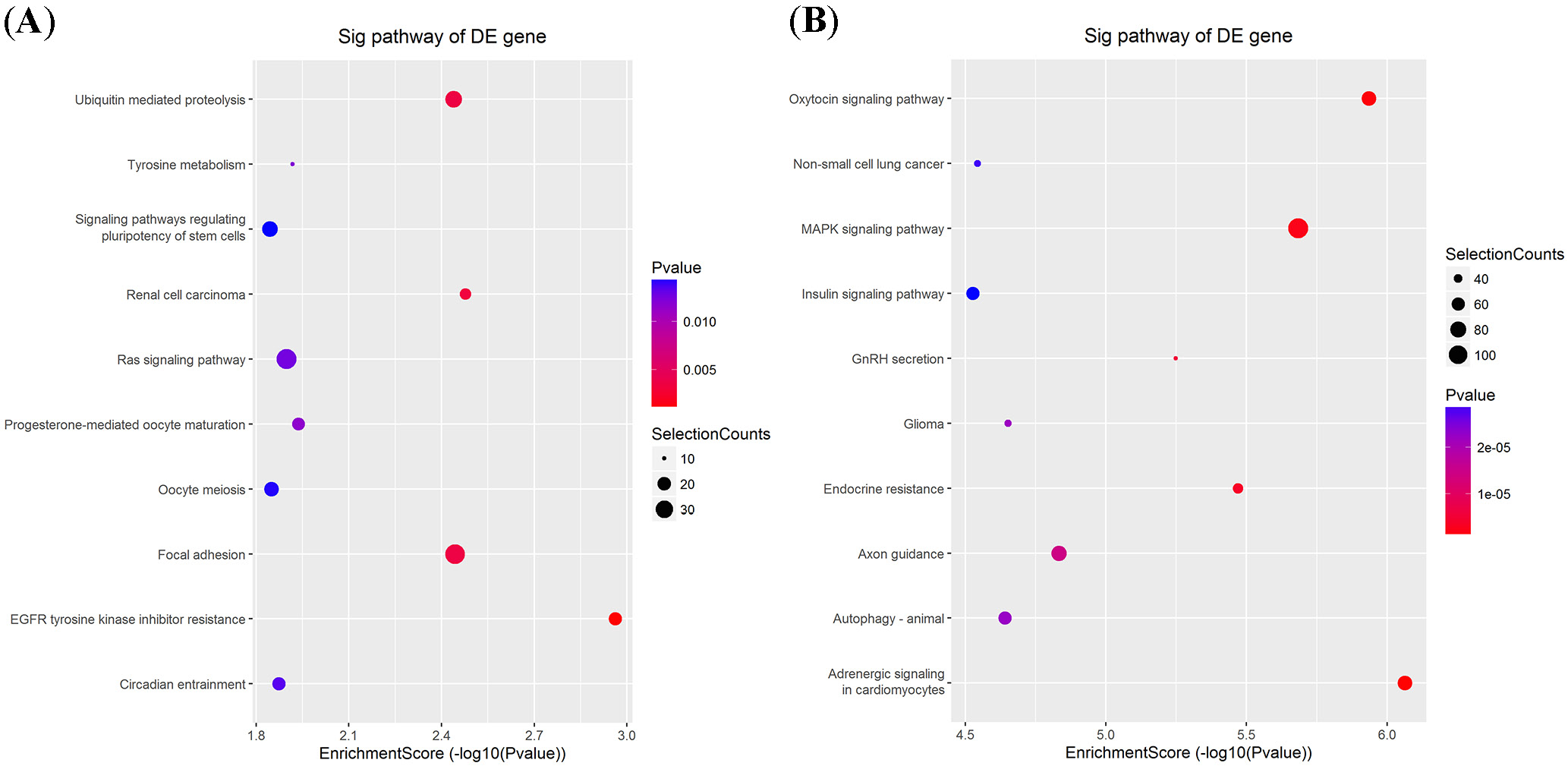
In contrast, the downregulated target genes were primarily enriched in primary metabolic processes and the establishment of localization (Figure 2D). In the cellular component category, they were enriched in membrane-bounded organelles and membranes (Figure 2E), while molecular functions were enriched in cation binding and heterocyclic compound binding (Figure 2F). The KEGG pathway analysis further revealed that 39 pathways were associated with the upregulated mRNAs and 127 pathways with the downregulated mRNAs. Notably, several crucial pathways, such as the insulin signaling pathway, Ras signaling pathway, MAPK signaling pathway, and autophagy, were potentially influenced by the altered circRNAs (Figure 3).
qRT-PCR confirmation of circRNAs’ differential expression
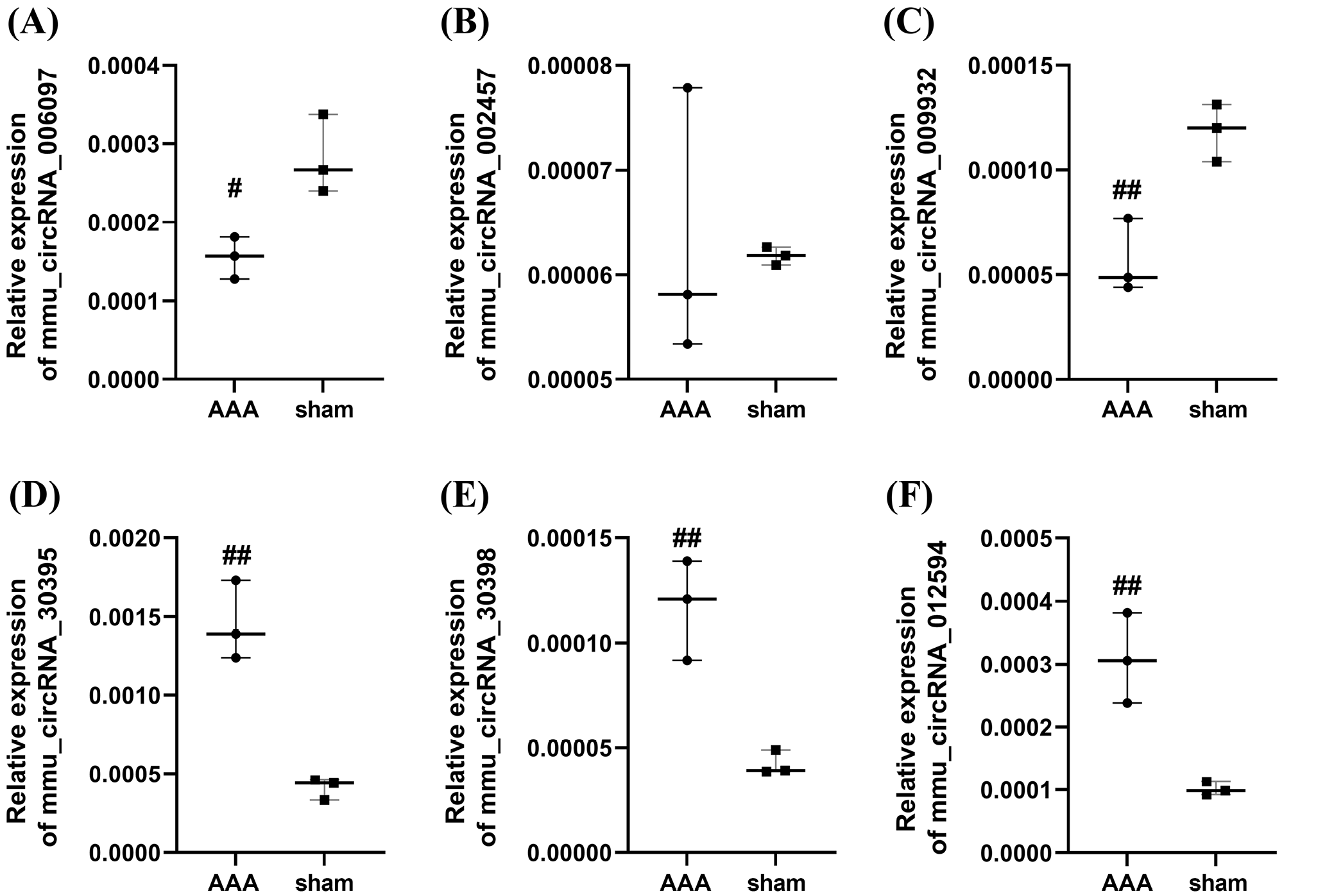
To validate the microarray results, 6 notably altered circRNAs, including circRNA_006097, circRNA_002457, circRNA_009932, circRNA_30395, circRNA_30398, and circRNA_012594, were assessed using RT-qPCR (Figure 4). The results demonstrated that the expression levels of cir- cRNA_006097 and circRNA_009932 were markedly downregulated (Table 1, Table 2, Supplementary Tables 2–5). Conversely, circRNA_30395, circRNA_30398 and circRNA_012594 were significantly upregulated in the AAA group (Table 3, Table 4, Table 5, Supplementary Tables 6–11). Conversely, circRNA_006097 and circRNA_009932 were markedly downregulated (Table 1, Table 2, Supplementary Tables 2–5). In contrast, circRNA_002457 showed no significant difference in expression between the AAA and control groups (Table 6, Supplementary Table 12,13) Overall, the RT-qPCR results were consistent with the microarray data, supporting the reliability of the expression profiles obtained from the circRNA microarray analysis.
Competing endogenous RNA (ceRNA) network construction
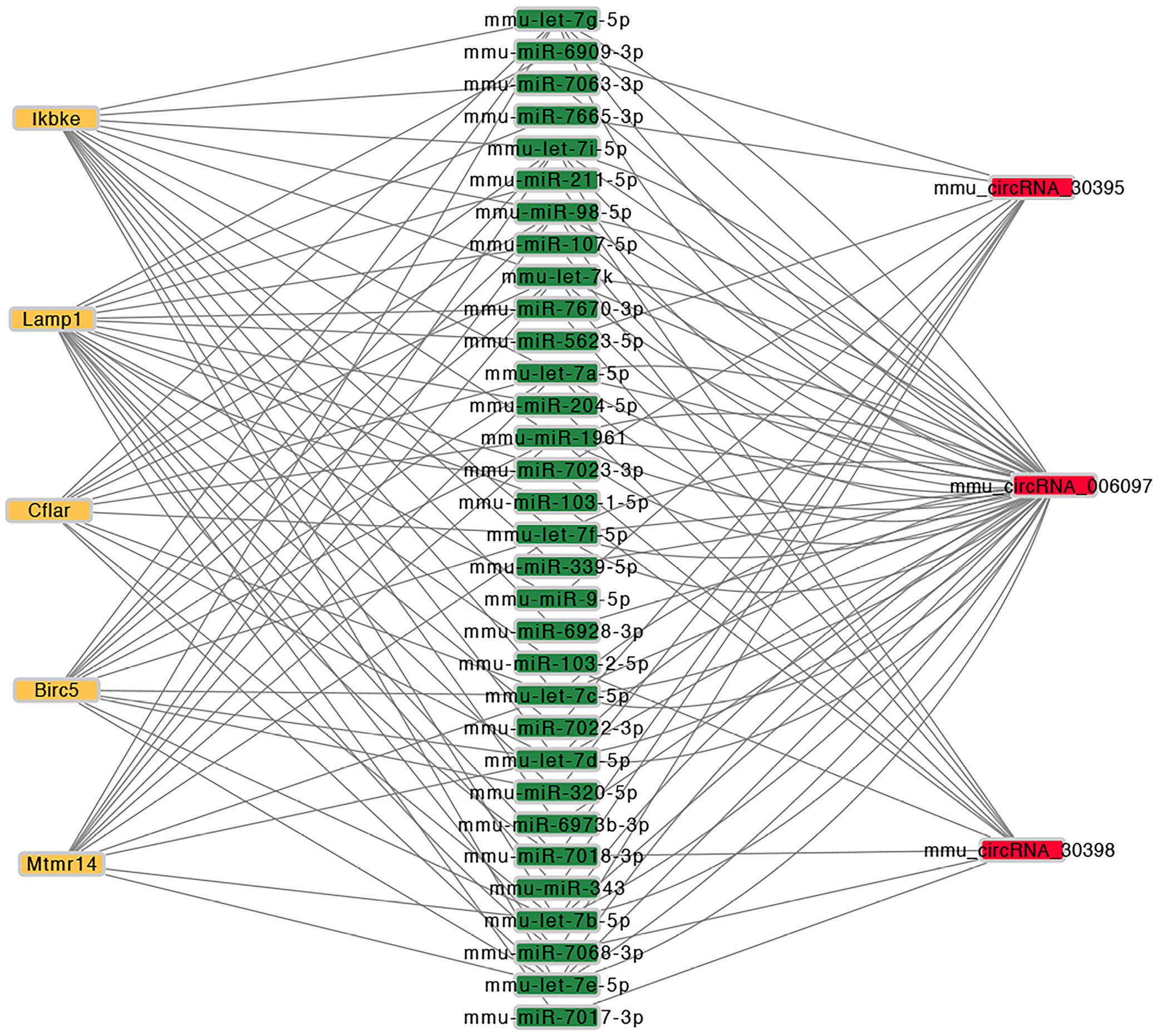
To further elucidate the potential regulatory mechanisms of the validated circRNAs, a circRNA–miRNA–mRNA interaction network was constructed using Cytoscape software (https://cytoscape.org) (Figure 5). This network was based on predicted miRNA binding relationships and included the top 5 miRNA candidates associated with each confirmed circRNA, as summarized in Supplementary Table 14.
Discussion
Advancements in RNA-sequencing technologies have revealed that circRNAs constitute a distinct class of non-coding RNA molecules expressed across a wide range of mammalian tissues.21 Recent studies have highlighted their potential roles as both therapeutic agents and disease biomarkers in various pathological conditions.22 Acting as miRNA sponges, circRNAs can modulate post-transcriptional gene regulation by preventing miRNAs from binding to their target mRNAs. In addition to their interactions with RNA molecules, circRNAs are also capable of binding specific proteins, thereby influencing their localization or activity. Moreover, some circRNAs regulate transcription within the nucleus, serve as templates for translation, and even compete with linear mRNA splicing.23 Due to their covalently closed-loop structure, circRNAs exhibit enhanced stability compared to linear RNAs, further supporting their potential as circulating biomarkers. Accumulating evidence suggests that circRNAs play important roles in cardiovascular biology and disease.22, 24
Given the absence of effective pharmacological treatments for AAA, surgical interventions (either open or endovascular) remain the only available options. Developing effective medical therapies for small AAAs or for patients unsuitable for AAA repair would significantly enhance the existing approach to patient management. In this study, we established an AAA mouse model through continuous subcutaneous infusion of Ang II in apoE–/– mice and systemically profiled circRNA expression in the aorta using circRNA microarray technology. Ninety circRNAs were found to be upregulated and 234 downregulated in the AAA group compared with controls. The predicted target genes of these differentially expressed circRNAs were functionally annotated through GO and KEGG pathway enrichment analyses.
Gene Ontology analysis revealed the involvement of differentially expressed circRNAs in diverse biological processes, such as nitrogen compound metabolism and the establishment of localization. Meanwhile, KEGG analysis highlighted enrichment in key pathways, including the MAPK, Ras, insulin signaling, and autophagy pathways. Among these, the MAPK signaling pathway was notably associated with the upregulated circRNAs in AAA. Jun amino-terminal kinases (JNK1/2/3), a major kinase in the MAPK family, have been implicated in AAA formation. Previous studies demonstrated that JNK inhibition attenuates AAA formation induced by Ang II or calcium chloride in mouse models.25, 26 Additionally, extracellular signal-regulated kinases (ERK), another key kinase in the MAPK family, have been shown to be elevated in human AAA tissues, as reported by Groeneveld et al.27
Autophagy, a critical cellular mechanism for degrading dysfunctional organelles and proteins, contributes to cellular homeostasis28 and has been increasingly linked to AAA pathogenesis.29 Associations between autophagy and established AAA risk factors, such as senescence,30 gender31 and cigarette smoking,32 have been documented. Furthermore, both pharmacological agents33, 34 and genetic interventions35, 36 targeting autophagy have shown efficacy in small animal models of AAA. In the current study, 6 candidate circRNAs were selected for RT-qPCR verification. The results demonstrated that the expression levels of mmu_circRNA_30395, mmu_circRNA_30398, mmu_circRNA_012594, mmu_circRNA_006097, and mmu_circRNA_009932 were significantly dysregulated, in accordance with the microarray analysis.
To identify the most informative candidates, the 5 validated circRNAs were used to construct a comprehensive ceRNA regulatory network. As mentioned earlier, autophagy plays a significant role in AAA pathogenesis, so we focused on autophagy-related target genes. Further bioinformatics analysis revealed that the target genes of mmu_circRNA_30398, mmu_circRNA_006097 and mmu_circRNA_30395 were predominantly enriched in the autophagy pathway. The downstream target genes of circRNAs involved in autophagy in this ceRNA network include Birc5, Cflar, Ikbke, Mtmr14, and Lamp1.
Further inspection of the ceRNA network revealed additional axes likely involved in AAA progression. Matrix metalloproteinases (MMPs), particularly MMP-2, are known contributors to extracellular matrix (ECM) degradation37 and AAA pathogenesis.38 Elevated MMP-2 activity in the aorta has been linked to Ang II and CaCl2 stimulation.39 Notably, Liu et al. reported that circ_MMP-2 promotes hepatocellular carcinoma metastasis by sponging miR-136-5p, resulting in MMP-2 upregulation.40 Our analysis identified miR-136-5p as a likely target miRNA of circRNA_30398, supporting the hypothesis that mmu_circRNA_30398 may facilitate AAA progression via the circRNA_30398/miR-136-5p/MMP-2 axis.
Additionally, miR-448-3p, miR-337-3p and miR-432 were predicted as potential targets of circRNA_006097. Notably, miR-448-3p has been shown to be dysregulated in intracranial aneurysms, suggesting a role in vascular pathogenesis.41 The AAA development is also closely linked to VSMC apoptosis and phenotypic changes, processes in which circRNAs are increasingly recognized as regulators.42 Structural deterioration of the arterial wall, including ECM degradation and VSMC loss, is a primary cause of aortic dilation and rupture. Several studies have highlighted the involvement of circRNAs in VSMC apoptosis and proliferation.43, 44 For instance, miR-432 has been shown to suppress PI3K/AKT/mTOR signaling by downregulating P55PIK, inhibiting myoblast proliferation and differentiation.45
Similarly, miR-337-3p modulates myosin 10 expression, impacting pulmonary artery smooth muscle cell proliferation.46 These findings suggest that circRNA_006097 may influence AAA formation by regulating VSMC function.47 Moreover, circRNA_009932 may exert similar effects through its predicted target, miR-222-5p, which inhibits smooth muscle cell differentiation by suppressing ROCK2 and α-smooth muscle actin expression.
Limitations
This study has several limitations. First, the construction of the circRNA–miRNA–mRNA interaction network was based solely on bioinformatics analysis, which requires further experimental validation. Second, although RT-qPCR confirmed the differential expression of selected circRNAs, their functional roles in AAA pathogenesis were not assessed through knockdown or overexpression experiments. Such mechanistic investigations are essential to establish causal relationships. Finally, emerging evidence suggests that circRNAs may also interact with RNA-binding proteins or serve as templates for translation, aspects that were not explored in the current study.
Conclusions
In this study, circRNA expression profiles in AAA were characterized based on an Ang II-induced mouse model. Five circRNAs were selected and verified with RT-qPCR. Our findings provide evidence that the circRNA-miRNA-mRNA axis may play a critical role in the molecular pathogenesis of AAA. Further experimental research is needed to elucidate the regulatory mechanisms of the circRNA_30398/miR-136-5p/MMP-2 axis. Understanding these mechanisms could reveal potential therapeutic targets for AAA.
Supplementary data
The supplementary materials are available at https://doi.org/10.5281/zenodo.15744894. The package includes the following files:
Supplementary Table 1. Primers used for RT-qPCR.
Supplementary Table 2. Shapiro–Wilk test results for circRNA_006097 mRNA expression in the AAA and sham groups (see Fig. 4A).
Supplementary Table 3. F-test for equality of variances for circRNA_006097 mRNA expression in the AAA and sham groups (see Fig. 4A).
Supplementary Table 4. Shapiro–Wilk test results for circRNA_009932 mRNA expression in the AAA and sham groups (see Fig. 4C).
Supplementary Table 5. F-test for equality of variances for circRNA_009932 mRNA expression in the AAA and sham groups (see Fig. 4C).
Supplementary Table 6. Shapiro–Wilk test results for circRNA_30395 mRNA expression in the AAA and sham groups (see Fig. 4D).
Supplementary Table 7. F-test for equality of variances for circRNA_30395 mRNA expression in the AAA and sham groups (see Fig. 4D).
Supplementary Table 8. Shapiro–Wilk test results for circRNA_30398 mRNA expression in the AAA and sham groups (see Fig. 4E).
Supplementary Table 9. F-test for equality of variances for circRNA_30398 mRNA expression in the AAA and sham groups (see Fig. 4E).
Supplementary Table 10. Shapiro–Wilk test results for circRNA_012594 mRNA expression in the AAA and sham groups (see Fig. 4F).
Supplementary Table 11. F-test for equality of variances for circRNA_012594 mRNA expression in the AAA and sham groups (see Fig. 4F).
Supplementary Table 12. Shapiro–Wilk test results for circRNA_002457 mRNA expression in the AAA and sham groups (see Fig. 4B).
Supplementary Table 13. F-test for equality of variances for circRNA_002457 mRNA expression in the AAA and sham groups (see Fig. 4B).
Supplementary Table 14. The top 5 predicted targets of RT-qPCR confirmed dysregulated circRNAs.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.