Abstract
Hematological malignancies encompass a diverse group of cancers affecting the blood, bone marrow and lymph nodes. The p16 gene, encoding the P16INK4A protein, plays a pivotal role in cell cycle regulation and tumor suppression. Understanding the involvement of p16 in the development and progression of hematological malignancies is crucial for advancing therapeutic strategies.
This systematic review aims to elucidate the multifaceted roles of the p16 gene and P16INK4A protein in hematological malignancies, focusing on their impact on disease pathogenesis, prognostic significance and therapeutic implications. A comprehensive search was conducted across electronic databases, including PubMed, Scopus and Google Scholar, using predefined search terms related to p16, P16INK4A, hematological malignancies, and therapy. Studies published up to 2023 were included, encompassing clinical trials, observational studies, meta-analyses, and preclinical research.
The review synthesizes evidence highlighting the dysregulation of the p16 pathway in various hematological cancers. Alterations in p16 expression levels, genetic mutations and epigenetic modifications contribute to disease initiation and progression. Moreover, the prognostic significance of p16 status in predicting therapeutic outcomes and patient survival is explored. The p16 gene and P16INK4A protein emerge as promising biomarkers and therapeutic targets in hematological malignancies. Integrating knowledge of p16 dysregulation into clinical practice holds the potential to optimize treatment strategies, enhance patient outcomes and pave the way for personalized medicine approaches in the management of these challenging diseases.
Key words: prognostic factor, hematological malignancies, p16, therapeutic implications, P16INK4A
Introduction
Hematological malignancies account for 9.7% of all cancers, ranking them the 4th most common in the human population.1 The huge number of cases poses a significant obstacle for the healthcare system. It is believed that a comprehensive understanding of cancer pathophysiology can serve as a basis for the discovery of new medications to optimize and maximize the outcomes of patients with hematological malignancies. Moreover, gaining further knowledge of the systems responsible for cancer formation and progression could potentially enable the early detection of susceptible patients and the implementation of preventive measures. Identification of predictive and prognostic indicators is crucial for tailoring preventive strategies, improving clinical practice and allocating research resources.2
P16INK4a is a protein classified as a tumor suppressor, mostly due to the widespread genetic inactivation of the p16INK4a (or CDKN2A) gene in almost all forms of human malignancies. P16INK4a is a component in regulating the cell cycle and working in conjunction with the P53 protein; research has demonstrated that an increased amount of P16INK4a expression (upregulation) has a role in cellular senescence, aging and the advancement of cancer. This suggests that the control of P16INK4a is significant for its proper functioning.3
The systematic review used PubMed, Scopus and Google Scholar search engines. The following key words were used: “p16INK4a”, “INK4a, P16INK4a”, “hematologic malignancies”, “acute myeloid leukemia”, “acute lymphoblastic leukemia”, “myelodysplastic syndromes”, “chronic myeloid leukemia”, “mastocytosis”, “chronic lymphocytic leukemia”, “Hodgkin’s lymphoma”, “Hodgkin’s disease”, “non-Hodgkin’s lymphoma”, “plasma cell myeloma”, “multiple myeloma”, and “prognostic factors”, “p16INK4a expression”, “DNA methylation”, “chromosomal deletions”. The search was limited to the period from 1993 to 2023. Articles published more than 10 years ago were used only when necessary. The sources were last searched on July 24, 2024.
Objectives
The primary objective of this systematic review was to present an overview of the existing knowledge on the function of the p16INK4a gene and P16INK4a protein in the development of hematological malignancies. Additionally, the review aimed to investigate the p16 gene and P16INK4a protein potential roles as prognostic factors, and the therapeutic implications of targeting the p16 gene and P16INK4a protein in anticancer treatment. Furthermore, the review attempted to familiarize the reader with the structure of these elements and the mechanisms of their interaction with the cell cycle.
Structure and function of the p16 gene and P16INK4a protein
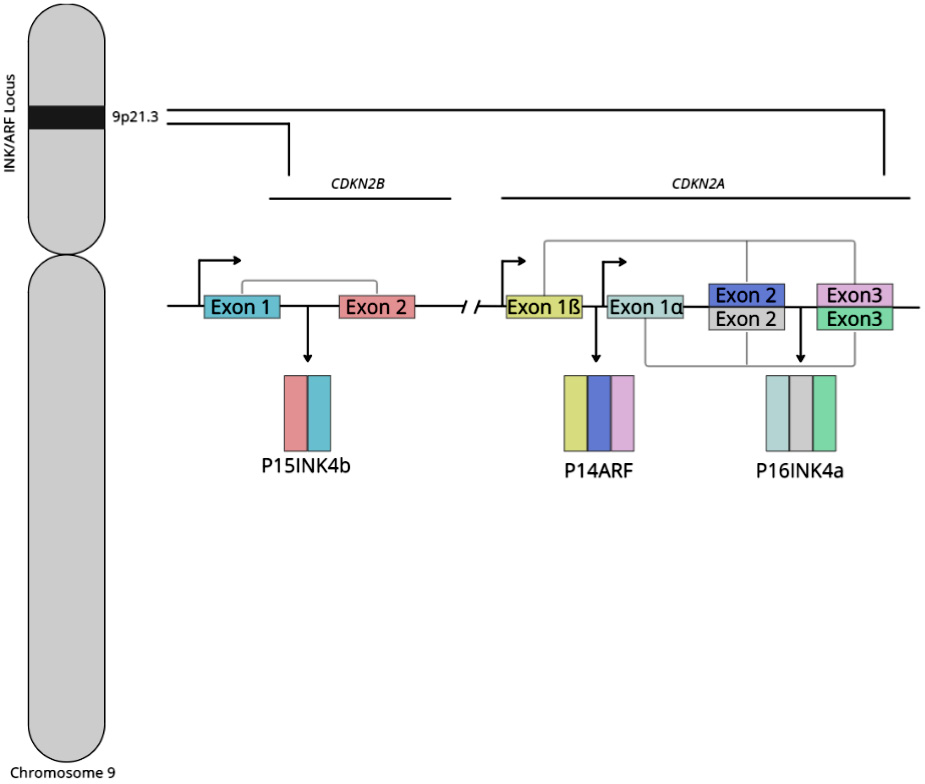
Serrano et al. identified P16INK4a as a protein that binds to CDK4 and inhibits the catalytic activity of the CDK4/cyclin D enzymes in 1993.4 The protein is also known as P16 or MTS1 (multiple tumor suppressor 1) and is encoded by the CDKN2A gene located at the INK4b/ARF/INK4a locus on chromosome 9p21.3 (Figure 1).5, 6 CDKN2A also encodes for P14ARF (P19ARF in mice), while CDKN2B encodes for P15INK4b, CDKN2C for P18INK4d and CDKN2D for P19INK4d.7 Additionally, the INK4b/ARF/INK4a locus encodes antisense non-coding RNA in the INK4/ARF locus (ANRIL).8
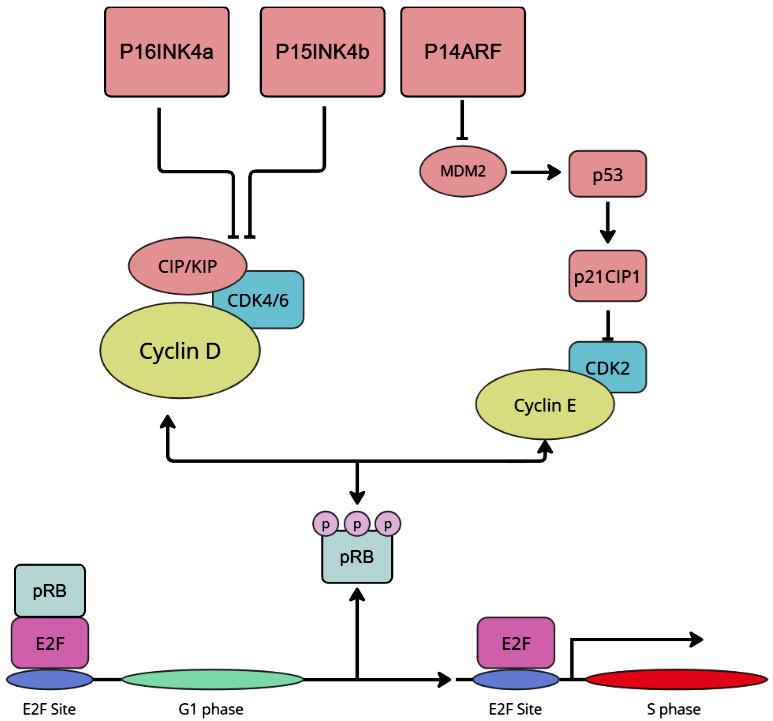
P16INK4a and P15INK4b bind and inhibit CDK4 and CDK6, which in turn are significant to relieve the cell cycle inhibitory activity of the retinoblastoma (Rb) tumor suppressor, whereas P14ARF is an activator of tumor suppressor p53 (Figure 2).7, 9, 10
P16INK4a and P15INK4b, as well as P18 and P19, belong to the INK4 protein family.9 The common feature of these 4 proteins is that they are built of ankyrin repeat (AR) motifs, each of which is composed of 31–34 amino acids.11 Ankyrin repeat motifs are found in many proteins and are responsible for protein–protein interactions. The studies have shown that P16INK4a and P15INK4b proteins are composed of 4 ankyrin repeat (AR) motifs, whereas P18 and P19 contain 5 AR motifs.12 In addition to their similar structure, all INK4 proteins bind to CDK4/6 and inhibit its activity. Unlike P16INK4a and P15INK4b, which are encoded by the same locus and most likely resulted from gene duplication,13 genes encoding P18 and P19 proteins are located within chromosomes 1p32 and 19p13.2, respectively.14
The studies on interactions between P16INK4a and CDK4/6 have shown that residues of P16INK4a that interact with CDK4/6 are located in the 2nd and 3rd AR motif. Crystallographic studies revealed that AR motifs present in the P16INK4a protein form a helix bundle with a concave surface in which clusters of charged groups are present for target binding.15
P16INK4a inhibits CDK4/CDK6 proteins in 2 ways. First, the concave surface of the P16INK4a protein interacts with a catalytic site of CDK4/6 proteins, leading to a decrease in the kinase activity of CDK4/6 proteins. Second, the interaction between P16INK4a and CDK4/6 induces changes in the tertiary structure of CDK4/6, which affects the binding between CDK4/6 and its activator, cyclin D.16
The studies conducted until now have revealed that the primary function of the P16INK4a protein is regulation of the cell cycle. P16INK4a involvement in the regulation of cell cycle progression occurs through at least 2 mechanisms. The best description is the contribution of P16INK4a to the pRb/E2F pathway. P16INK4a binds and inhibits the activity of CDK4 and CDK6 proteins, whose role is to phosphorylate Rb proteins. When P16INK4a inhibits the activity of the CDK4/6 protein, it results in hypophosphorylation of the Rb protein. Hypophosphorylated Rb protein inhibits association of E2F protein with promoters of proliferation-associated genes (e.g., cyclin B1, dihydrofolate reductase, thymidine kinase, and JunB proto-oncogene), thus preventing the entrance of cells into the S phase and proliferation. On the other hand, cell commitment to proliferation results in pRb hyperphosphorylation by CDK6/4 in late G1, resulting in the entry of the cell into the S phase (Figure 2). The 2nd mechanism involves the interaction of P16INK4a with TFIIH in the pre-initiation complex, which prevents the CDK7 subunit of this transcription factor from phosphorylating the carboxy-terminal domain of the large subunit of RNA polymerase II, a step required for the transcription of RNAII polymerase-dependent genes.17, 18
Regulation of P16INK4a expression
Traversing the G1/S cell cycle checkpoint is one of the critical events in cell life and is therefore tightly regulated by multiple pathways. In general, the main outcome of this regulation should be to allow only fit and unstressed cells to proliferate and to inhibit the proliferation of cells exposed to stress and oncogenic stimuli. In general – because of the importance of the p16INK4a gene for cell cycle control – this gene is maintained in a repressed state in normal cells so they can proliferate. Expression of this gene requires: 1) a permissive state of chromatin surrounding the p16INK4a gene; and 2) stimulation of transcription by activator proteins. The induction of p16INK4a gene expression is usually observed during cellular senescence, a state of the cell during which the cell is metabolically active but cannot proliferate.12 The cellular senescence might be induced by a number of factors, including UV light, reactive oxygen species (ROS) and oncogenes.19
Epigenetic regulation
It has been reported that the p16INK4a gene undergoes in cells silencing by evolutionary conserved multiprotein complexes polycomb repressive complex 1 (PRC1) and polycomb repressive complex 2 (PRC2).5 The studies have shown that long non-coding (Inc) RNAs such as ANRIL and MOV10 are responsible for the recruitment of the PRC2 complex to the p16INK4a gene.20 According to the well-established model, the enzymatic subunits of this complex, Ezh1 and Ezh2, are responsible for the trimethylation of lysine 27 of histone H3 (H3K27me2/3),21 which is next recognized by the subunit of the PRC1 complex, the CBX protein, and the entire PRC1 complex is recruited to target locus.22 A part of the PRC1 complex E3 ubiquitin ligase RING1A/B is responsible for the monoubiquitination of histone H2A at lysine 119 (H2AK119ub1), which, along with local chromatin compaction, leads to gene silencing. The studies also showed that histone deacetylases (HDAC) are involved in INK4 genes silencing. One mechanism by which HDACs contribute to this phenomenon is that they promote dissociation of E2F1 from Rb, and the former induces transcription of C-MYC, which in turn is a positive regulator of BMI-1 expression. BMI-1 is a part of the PRC1 complex.23 Similarly, Ezh protein levels are increased as a result of E2F dependent transcription, leading to increased activity of the PRC2 complex.5 This altogether results in long-term reversible suppression of the INK4 genes: p19ARF and p16INK4a. The mechanism responsible for the removal of repressive histone marks from the p16INK4a gene is less understood; however, the analyzed studies point to several possible mechanisms. First, they suggest the involvement of histone demethylases. In particular, levels of H3K27me3 demethylase Jmdjd3 were shown to be increased by oncogenic stressors, leading to the removal of repressive histone marks from the p16INK4a gene promoter,24 resulting in dissociation of the PRC1 complex, and decondensation of p16INK4a gene promoter. Second, Jun dimerization protein 2 (JDP2) may bind and sequester H3K27 away from PRC2.19
In addition to this reversible repression of the p16INK4a gene locus, PRC1 and PRC2 complexes are responsible for irreversible p16INK4a gene silencing that occurs in many forms of cancer. Irreversible repression of the p16INK4a locus is characterized by trimethylation of H3 at lysine 9 by the PRC2 complex, binding of the HP-1 adaptor molecule, and irreversible binding of the PRC1 complex.25
Regulation of p16INK4a gene transcription
The expression of the p16INK4a gene can be regulated by several transcription factors.23 It should be noted that there is complex interplay between positive and negative regulators of p16INK4a gene transcription that allows to fine-tune p16INK4a gene expression in response to multiple signals to which cells are exposed – e.g., oncogenic signaling leads to the activation of Ras/MEK/MAP kinase. This pathway may lead to increased activity of at least 3 transcription factors that stimulate p16INK4a gene expression. One of them is an HMG box containing protein 1 (HBP1). It was shown that HBP1-induced expression of the p16INK4a gene may lead to cellular senescence.26
It is also well established that this signal stimulates increased activity of Ets1 or Ets2 transcription factors. These proteins recognize the E-box element located in the p16INK4a gene and stimulate its transcription. It is worth noting that other signaling pathways may modulate Ets1/2 transcriptional activity. For example, inhibitor of DNA binding 1 (ID1) prevents the binding of Ets1/2 to the p16INK4a gene promoter.27
Another example of fine-tuning p16INK4a gene expression is the interaction of AP1 transcription factors with the p16INK4a gene promoter. AP-1 transcription factors are known to regulate gene expression in response to a variety of signals, including cytokines, growth factors, stress signals, bacterial and viral infections, and oncogenic stimuli.28 The overexpression of certain proteins, e.g., Jun-B in cells, leads to increased P16INK4a levels and the premature senescence in primary mouse fibroblast.26 In contrast, another AP1 protein, c-Jun, reduces the expression of p16INK4a, promoting cell proliferation.29 It is worth noting that the P16INK4a protein inhibits c-Jun kinase, which is necessary for c-Jun activation and oncogenic transformation induced by ultraviolet (UV) light.30
The role of the P16 gene and P16INK4a protein in hematological malignancies
In many human cancers, the INK4b/ARF/INK4a locus on chromosome 9p21 is completely deleted. Additionally, inactivation of p16 at the gene level is observed and may include homozygous deletion, promoter hypermethylation, loss of heterozygosity (LOH), or point mutation. For example, 48% of pancreatic cancer samples contain homozygous deletions of the p16INK4a gene, and approx. 73% of hepatocellular carcinoma samples have hypermethylation of the p16 promoter. While deletions of the p16 gene and promoter hypermethylation lead to complete loss of the P16 protein, mutations can have variable effects on the activity and function of this protein.3
It has been hypothesized that p16INK4a serves as a backup tumor suppressor gene for p53. In normal cells where p53 signaling is unaltered, oncogenic Ras stimulation triggers DNA damage, which in turn leads to activation of p53, induction of p21, and inhibition of cell proliferation. In numerous cancerous cells, p53 is rendered nonfunctional, potentially resulting in DNA damage due to hyperproliferation. In such cells, DNA damage may trigger the expression of the p16INK4a gene and the onset of cellular senescence. However, when both the p53 and p16INK4a genes are inoperative, this condition facilitates the unchecked proliferation of cancer cells.31
Given the prevalence and mortality of hematological malignancies, especially among older individuals with multiple comorbidities, investigators continuously seek new predictive and prognostic markers. A thorough evaluation of a patient’s health condition and prognosis before treatment can assist physicians in optimizing and tailoring therapy, ultimately leading to favorable treatment outcomes.32
In the following sections, we will examine existing data on the significance of the p16INK4a gene and the P16INK4a protein as prognostic factors in hematological malignancies. We will explore their involvement in tailoring treatments to individualize and personalize anticancer therapy. Additionally, we will assess the potential influence of deletions, mutations and other alterations of the p16 gene and P16INK4a protein on the prognosis of patients with hematological malignancies.
Acute myeloid leukemia
Acute myeloid leukemia (AML) is characterized by distinct biological and clinical pathways in patients of different age groups, with older patients often exhibiting altered gene expression and karyotype abnormalities. The highest incidence of AML is observed in older indviduals, which may be related to the expression of p16INK4a mRNA in bone marrow stromal cells.33 Although AML patients often experience remissions following therapy, overall survival (OS) rates remain unsatisfactory.34 The reduced long-term survival rate is primarily due to weaker cytostatic tolerance and therapy resistance in older patients.33
Abdul-Aziz et al. showed that high levels of the p16INK4a gene in bone marrow stem cells are linked to the presence of pro-tumor factors related to senescence-associated secretory phenotype (SASP) and the promotion of the survival of AML cancer cells. Removing p16INK4a mRNA stops its effect and slows the growth of the tumor.34
Tschan et al. quantitatively assessed the level of p16INK4a and p14ARF mRNA expression. Higher amounts of p16INK4a and p14ARF were found in myeloblasts compared to normal monocytes and granulocytes, according to their study. The obtained results may suggest that increased expression levels of both of these genes participate in the pathogenesis of AML.35 De Jonge et al. conducted a study of p16INK4a mRNA expression to identify molecular differences between older and younger AML patients. They concluded that the expression of p16INK4a mRNA in older individuals, belonging to the intermediate- and high-risk groups, is much lower than in the group of young patients, and the p16INK4a gene may be an independent prognostic factor.33
Faderl et al. analyzed the deletion of p16INK4a, p14ARF and p15INK4b in 74 patients with AML. Additionally, to determine the phosphorylation of Rb protein in patients with deletions of p16INK4a, p14ARF and p15INK4b, the western blot method was utilized. Deletions of p16INK4a, p14ARF and p15INK4b occurred in 5% of the patients in the studied group, indicating that such chromosomal aberrations occur at a low frequency in patients with AML. The study also compared the complete response (CR) rate of the disease, duration of CR and OS of patients without deletions of p16INK4a, p14ARF and p15INK4b with those with the aforementioned abnormalities. The results suggest that patients with deletions of p16INK4a, p14ARF and p15INK4b have significantly shorter duration of CR and OS rates compared to patients without deletions.36
Acute lymphoblastic leukemia
Radiotherapy, allogeneic hematopoietic stem cell transplantation (allo-HSCT), multidrug treatment, and a better understanding of chromosomal translocations and somatic mutations have improved OS in acute lymphoblastic leukemia (ALL) patients.37, 38 Young patients and children show significant progress. Prognosis for older patients is still unsatisfactory. This is because chromosomal, clinical and biochemical issues are more frequent. The poor prognosis of older patients is also due to treatment-related mortality and/or less toxic but less effective treatment regimens.39 Prognostic variables for adult ALL patients include age, leukocyte count and cytogenetic abnormalities. Disease risk assessment must be enhanced due to high treatment failures.40
Stock et al. examined lymphoblasts for abnormalities of the p16INK4a, Rb and p53 genes. Disorders of the p16INK4a gene were found in 17 patients. The mutation occurred in 7 patients: 6 with the Rb mutation and 3 with the p53 mutation. All 3 changes occurred in 1 patient. Then, in 5 patients with a detected p16 gene mutation, CD34+ Lin- cells were isolated and immunohistochemical examination was performed. In 3 patients, there was a complete lack of p16INK4a expression; in the remaining 2 patients, the expression was reduced. This may indicate that mutations leading to the loss of p16 expression may be a step in the leukemogenesis of ALL.40
Omura-Minamisawa et al. proposed that knocking down the p16INK4a gene at the RNA or protein level causes T-cell acute lymphoblastic leukemia (T-ALL). Previous investigations showed DNA loss, mutation or hypermethylation inactivating p16INK4a. Authors observed that 93% of T-ALL patients had p16INK4a switched off at the DNA, RNA and protein levels. This shows that T-ALL pathogenesis requires p16INK4a inactivation.41
Fizzotti et al. examined DNA samples from patients with hematological malignancies to determine the frequency of homozygous p16 gene deletion. In 10 of 58 samples (17%) of B-ALL and T-ALL patients, p16 homozygous deletions were found. Homozygous p16 deletion patients had more leukocytes and leukemic cells than those with the normal gene. The authors also examined 2 cases of ALL in which DNA samples were available, collected before the start of treatment and after disease progression. In both cases, the occurrence of the p16 deletion was confirmed during relapse. Therefore, the authors concluded that deletion of the p16 gene may help identify patients with features of aggressive disease and may be a negative prognostic factor.42
Soenena et al. studied adult ALL patients’ P16INK4a protein expression. Patients with negative immunocytochemical tests had significantly worse OS and event-free survival (EFS). This was only found in standard-risk karyotype patients.43
Wang et al. researched the prognostic and clinical effects of p16 gene deletion in ALL patients. P16 deletions were found in 33 of 86 cases. Higher leukocyte and decreased platelet counts were associated with the p16 deletion. Treatment results for EFS and OS were significantly poorer with the p16 deletion. Allo-HSCT was performed on 22 of 33 p16 deletion patients. Patients with deletion had prolonged EFS and OS following allo-HSCT. The authors concluded that p16 deletion is a significant, negative prognostic factor in adult ALL, while performing allo-HSCT in this group of patients markedly improves the prognosis.44
Myelodysplastic syndromes
Myelodysplastic syndromes (MDS) is a group of hematopoietic stem cell disorders characterized by inefficient hematopoiesis and frequent transformation to AML.45 This disease occurs mainly in older individuals.46 Accelerated cell aging, oxidative stress and aberrant DNA methylation may induce MDS.47 Currently, many different therapies are used in MDS. However, the only option that guarantees a complete cure is allo-HSCT. Unfortunately, many patients cannot undergo this procedure due to advanced age or concomitant diseases. Therefore, the main goal in the treatment of patients with MDS who cannot undergo allo-HSCT is to prolong and improve quality of life. This is possible thanks to the use of optimized treatment adapted to the patient’s opportunities and prognosis.48
Wang. et al. conducted a study aimed at determining the impact of cell aging on the development and prognosis of patients with MDS. The results showed increased expression of p16INK4a mRNA and P16INK4a protein in cells collected from MDS patients compared to the control group. Interestingly, AML patients showed lower p16INK4a mRNA expression compared to samples collected from healthy volunteers and MDS patients. This may suggest that the increased expression of p16INK4a mRNA in the cells of MDS patients is associated with the occurrence of this disease.49
Other conclusions were presented by Papageorgiou et al., who examined p16 and p27 gene alterations in 51 untreated MDS patients. The Southern blot showed no homozygous p16 deletions. Except for 2 allelic polymorphisms, polymerase chain reaction-single-strand conformation polymorphism (PCR-SSCP) analysis and Sanger sequencing could not find p16 point mutations. The foregoing data imply that the p16 gene is not mutated during MDS pathogenesis and that other mechanisms boost expression.50
Nakamaki et al. studied CDK genes in MDS to see if genetic alterations in these genes affect MDS development and progression. The results showed no alterations in the p16 gene coding areas, suggesting that genetic anomalies of the gene are rare in MDS and cannot be used as a prognostic factor.51
Chronic myeloid leukemia
Chronic myeloid leukemia (CML) originates from stem cells. A characteristic genetic abnormality in this disease entity is the chimeric fusion gene BCR/ABL1, which arises from the rearrangement of the Philadelphia chromosome t(9;22)(q34;q11). The introduction of molecularly targeted medicines, tyrosine kinase inhibitors (TKIs), has drastically changed the method of treatment and risk assessment in CML. Currently, the most important prognostic factor in CML is the early response to treatment with TKIs.52 Every study on the p16 gene and P16INK4a protein as a predictive factor was published before TKIs were extensively used.
Lee et al. tested samples collected from healthy donors and patients with granulocytic reactions, and found barely detectable expression of p16INK4a mRNA. However, p16INK4a mRNA expression was abnormally increased in CML patient samples. P16INK4a overexpression in CML patients occurred much more often (80%) than in the control group.53
While studying CML patients’ p16INK4a suppressor gene expression, Cividin et al. noted other observations. In 11 untreated chronic CML patients, p16INK4a mRNA levels were low. Interferon alpha (IFN-α)-treated individuals, especially resistant ones, had significantly higher p16INK4a levels. This may show the p16INK4a gene’s predictive relevance in CML therapy.54 Güran et al. examined p53, p16INK4a, p15INK4b, and p57KIP2 gene mutations and homo/hemizygous deletions in interferon alpha (IFN-α)-treated CML patients. The study examined molecular changes linked to disease progression. Blast crisis occurred in 4 of the 12 study participants during therapy. All 4 instances had p53, p16INK4a and p15INK4b mutations. In the conclusions, the authors observed a correlation between the progression of CML and somatic mutations in p53, p16INK4a and p15INK4b, and suggested that detecting alterations in the genes p16INK4b and p15INK4b, as well as mutations in p53, may serve as prognostic markers in patients with CML.55
Xu et al. examined CML blast crises and homozygous p16 gene deletions. After conducting the analyses, no homozygous deletion of the p16 gene was found in the chronic phase. Deletion was found in samples collected from patients in myeloid blast crisis, in lymphoid blast crisis and in mixed blast crisis. The results suggest that there is a strong correlation between homozygous deletions of the p16 gene in lymphoid blast crisis, and conducting PCR testing may help in the early detection of CML patients at risk of blast crisis occurrence.56
Mastocytosis
Mastocytosis refers to a number of diseases defined by clonal mast cell proliferation in various organs. It can emerge at any age and take many forms. The somatic KIT D816V mutation affects 90% of adult patients.57
Tsujita et al. examined mastocytosis P16INK4a protein expression immunohistochemically. All 4 mastocytosis samples had P16INK4a protein overexpression. The scientists also reported decreased Ki-67 expression in cancer cells. The scientists found that overexpression of the P16INK4a protein ages cancer mast cells and suppresses their growth, reducing mastocytosis.58
A review of the currently available literature revealed that the role of the p16 gene and P16INK4a protein in the prognosis of mastocytosis has not been sufficiently investigated and described.
Chronic lymphocytic leukemia
In Western countries, the most common type of leukemia is chronic lymphocytic leukemia (CLL).59 The pathogenic molecular pathways of the disease are unknown.60 Patients with active, symptomatic disease or advanced stages of the disease require therapy. When treatment is necessary, there are several options, including targeted therapies. Clinical staging systems provide prognostic information based on physical examination results and blood counts. Early identification of patients who are likely to relapse appears to be the most effective way to avoid progression-related adverse events.61
According to Tsirigotis et al., CLL patients seldom have p16 gene mutations. In this study, a total of 34 samples from CLL patients were analyzed. The Southern blot analysis showed non-rearranged bands in 33 out of 34 cases. No homozygous deletions were detected in any of the cases.
The PCR-SSCP analysis of exons 1 and 3 showed a normal migration pattern in all examined cases.
In contrast, the analysis of exon 2 identified an abnormal migration pattern in 2 out of 34 cases.
The methylation analysis of the p16 gene promoter revealed hypermethylation of CpG islands in 6 out of 34 cases.60
Forsterová et al. examined 5 tumor suppressor gene promoter methylations. Abnormal p16 methylation was observed in 31% of patients. It suggests that the p16 gene is not important for determining the stage and prognosis of patients with CLL.62
Given the foregoing findings, p16 gene promoter methylation’s significance in CLL’s etiology requires conducting a larger number of studies.
Non-Hodgkin lymphoma
Non-Hodgkin lymphomas (NHLs) are lymphoid malignancies with distinct biochemical and clinical characteristics.63 Despite significant advances in therapeutic modalities, the lack of etiological data continues to impede the development of optimal treatment strategies for NHL. Several variables that may coexist in the incidence of these disorders have been explored, but the results were unclear.64
Pinyol et al. studied how p16INK4a gene alterations cause NHLs. The Southern blot was used to examine the p16 gene in 95 genomically available lymphomas, including 55 indolent and 40 aggressive cases. P16 homozygous deletions were found in 10 tumors. In 1 blastoid mantle cell lymphoma (MCL) without DNA, cytogenetic analysis found a 9p21 locus hemizygous deletion. Ultimately, the results suggest that p16 gene mutations are not common in NHLs.65
Fresh tissues from patients with untreated NHLs were tested for homozygous deletions and point mutations in the p16 coding areas by Gombart et al. One diffuse large B-cell lymphoma (DLBCL) cell line and 2 uncultured lymphomas: 1 large B-cell and 1 mixed T-cell lymphoma showed homozygous deletions of either the p16 gene or both the p15 and p16 genes. On the other hand, no point mutations were found in the lymphomas or cell lines. These results suggest that the rate of p15 and p16 gene alterations in lymphomas is low, but some lymphomas may develop as a result of p16 and/or p15 loss.66
Liu et al. analyzed NHL patient’s tumor samples collected before treatment. The samples were examined for p16 gene and P16INK4a protein expression abnormalities. Thirty-four of 64 samples had P16INK4a protein, while 45 had p16 mRNA. Results suggest that post-transcriptional abnormalities may contribute to NHL etiology in some cases.67
Villuendas et al. analyzed the expression of P16INK4a in various types of NHLs. Loss of P16INK4a expression was observed in 41 of the 112 samples tested. The loss of P16INK4a expression was observed in all samples that exhibited a high degree of malignancy. Loss of P16INK4a was found more frequently in cases progressing from low-grade lymphomas (e.g., follicular lymphoma) compared to high-grade lymphomas (e.g. DLBCL). The authors suggested that the loss of P16INK4a protein expression contributes to the progression of lymphomas, and gene expression level (assessed before starting treatment) may be a prognostic factor.68
Martinez-Delgado et al. examined lymphoid nodes and bone marrow samples for p16INK4a and p15INK4b methylation. All patients were tested for monoclonal immunoglobulin and T-cell receptor gene rearrangements. Six patients were diagnosed with gene methylation. Two patients showed methylation during therapy. Five instances were never methylated. Disease signs revealed methylation. This suggests that p16INK4a methylation may be a valuable disease marker and help detect recurrence.69
Hodgkin’s lymphoma
Hodgkin’s lymphoma (HL) is one of the most common lymphomas.70 A characteristic feature of HL and a factor necessary to make the diagnosis is the presence of Reed–Sternberg cells. These cells have a large cell morphology, and cell enlargement is one of the hallmarks of aging.71 Hodgkin’s lymphoma is considered a highly curable disease with first-line chemotherapy and/or radiotherapy. Unfortunately, some patients resist treatment or progress during or after the end of therapy. Numerous HL patients die prematurely due to the late toxic effects of intensive treatment.70
Although the same chemotherapy regimen (doxorubicin, bleomycin, vinblastine, and dacarbazine) has been the mainstay of treatment over the past 30 years, risk-adapted approaches have helped to de-escalate therapy in low-risk patients while intensifying treatment in high-risk patients.72
Garcia et al. conducted a study involving immunohistochemical staining of 40 samples collected from patients with HL to elucidate whether p16 inactivation is involved in the development of this disease entity. Polymerase chain reaction was also utilized to assess exon 1 methylation of the p16INK4a gene. Thirty of 37 instances showed a p16INK4a loss. Only 7 samples had tumor cell nuclear expression. The PCR analysis found gene hypermethylation in 14 of 23 instances. The p16INK4a gene was hypermethylated in 25% of pre-treatment samples and 83% of relapse samples. Methylation may be relevant for HL recurrence. The data also demonstrate that Reed–Sternberg cells lose p16INK4a gene expression, which may support the hypothesis that p16 is involved in HL.73
Calio et. al. examined 147 patients to investigate if the expression of the protein P16INK4a in HL may help clinicians predict EFS. Samples were tested for P16INK4a and P21CIP1/WAF protein levels. The findings revealed that both compounds may be independent EFS prognostic factors. The objective response rate (ORR) was 45% for patients with low P16INK4a expression and 89% for those with high expression.74
Plasma cell myeloma
Plasma cell myeloma (PCM) is characterized by infiltration of the bone marrow by malignant plasma cells, which synthesize and secrete monoclonal immunoglobulins (Ig) or their fragments75 Plasma cell myeloma mostly affects the elderly population.76 In the plasma cells of PCM, cytogenetic abnormalities are frequently detected, which serve as the basis for classifying patients into risk groups.77 It is known that abnormalities in genes controlling the cell cycle, including p16, contribute to the carcinogenesis of PCM.78 The ever-increasing number of medications used to treat PCM has improved the response and OS rates in patients with PCM. However, biomarkers for intensifying, de-escalating or completely changing treatment have not yet been established.79
According to Ng et al., up to 2/3 of studied PCM cells have hypermethylation that causes p16 gene function loss at diagnosis. Patients with plasmablastic transformation have 100% hypermethylated cells that silence the p16 gene. The results of the studies conducted using cell lines and primary PCM confirm that hypermethylation of p16 plays a significant role in PCM carcinogenesis.80
Wang et al. published a meta-analysis aiming to comprehensively assess the potential role of p16INK4a in the pathogenesis of PCM. The research results showed that the frequency of p16INK4a methylation was significantly higher in patients with PCM compared to healthy individuals.81
Chen et al. examined how often the p16 and p15 genes are methylated in PCM. In 10 of 22 instances, the p16 and p15 genes were methylated. According to the investigators, patients with p16 and p15 gene methylation experienced delayed cell apoptosis, an insufficient therapeutic response and a short OS. This suggests that methylation of the p16 and p15 genes is important for PCM apoptosis and prognosis.82
Mateos et al. examined the methylation status of the p16 gene in samples collected from patients with PCM. Forty-one samples showed methylation. Moreover, the percentage of S-phase plasma cells in these patients was almost 3 times higher than in patients with unmethylated p16 genes. The authors observed a correlation between the presence of p16 methylation, an increased level of B2-microglobulin and high values of C-reactive protein (CRP). Patients with the methylated p16 gene also had shorter OS and progression-free survival (PFS) rates. In results, the authors suggested that methylation of the p16 gene is a common event in PCM patients at the time of diagnosis and is associated with an increased rate of plasma cell proliferation and a worse prognosis.83
The potential therapeutic implications of targeting p16 gene and P16INK4a protein in hematological malignancies
The therapeutic potential of targeting the p16 gene and the P16INK4a protein in various cancers is highly significant.84, 85
Based on the data presented in the preceding sections of this review, it is plausible to hypothesize that therapies employed in the treatment of hematological malignancies (often characterized by disruptions in cell cycle regulation) that focus on reactivating or enhancing the expression of the p16 gene may restore proper cell cycle control and thereby contribute to the inhibition of cancer progression.86
The analyzed studies suggest that reactivating or increasing the expression of the p16 gene can be achieved through gene therapy or epigenetic modifications, such as reducing DNA methylation.87 For example, the use of demethylating agents or histone deacetylase (HDAC) inhibitors has shown promise in reactivating the expression of tumor suppressor genes, including those involved in key regulatory pathways disrupted in tumorigenesis.88, 89, 90, 91
Li and Seto have shown that HDAC inhibitors, such as suberoylanilide hydroxamic acid (SAHA), can induce the re-expression of silenced genes, including p16, thereby promoting cell cycle arrest and apoptosis in cancer cells.92
Furthermore, the combination of these epigenetic therapies with other treatment modalities has been explored. For instance, the integration of demethylating agents with traditional chemotherapy has shown enhanced efficacy in preclinical models of hematological malignancies.93, 94 Such combination therapies may potentiate the reactivation of P16INK4a, leading to improved therapeutic outcomes and offering a promising avenue for personalized cancer treatment strategies. Li et al. suggested that demethylation of the p16 gene may enhance the sensitivity of cancer cells to anti-cancer treatments, making cancer cells more susceptible to traditional chemotherapy and thereby rendering it a more effective treatment method.95
The P16INK4a protein plays a crucial role in cellular senescence, a state characterized by the irreversible cessation of cell division in previously proliferative cells. This process acts as a critical barrier to tumor progression by limiting the replicative potential of cancer cells. Employing therapeutic strategies to increase P16INK4a levels could, therefore, promote cellular senescence in cancer cells, effectively halting their proliferation and inducing a state resistant to further malignant transformation.7, 96
The potential of P16INK4a to induce senescence has been substantiated by several studies. For example, Alcorta et al. suggested that overexpression of P16INK4a in human fibroblasts leads to premature senescence, underscoring its role in cell cycle regulation and tumor suppression.97 Similarly, Collado et al., found that oncogene-induced senescence is often mediated by the upregulation of P16INK4a, highlighting its significance in tumor biology.98
Integrating therapies targeting the P16INK4a protein with other molecularly targeted drugs, such as TKIs or immune checkpoint inhibitors (CPIs), holds significant potential for achieving synergistic effects and thereby improving overall treatment efficacy. The P16INK4a protein, a critical tumor suppressor, inhibits cyclin-dependent kinases 4 and 6 (CDK4/6), which play a pivotal role in regulating the cell cycle. By targeting these mechanisms, combination therapies can simultaneously disrupt multiple pathways essential for cancer cell survival, enhancing therapeutic outcomes. Preclinical and clinical studies have demonstrated that CDK4/6 inhibitors, such as palbociclib, ribociclib and abemaciclib, show promising results in treating various cancers, particularly those characterized by dysfunction of the tumor suppressor P16INK4a. These inhibitors work by halting cell cycle progression, which is particularly effective in cancers with disrupted cell cycle regulation. Such cancers include not only solid tumors like breast cancer but also hematological malignancies, where aberrations in cell cycle regulators are common.99, 100, 101, 102, 103
For instance, the combination of the CDK4/6 inhibitor palbociclib with endocrine therapy has shown improved outcomes in hormone receptor-positive breast cancer. In a study by Finn et. al., patients receiving palbociclib in combination with letrozole exhibited significantly prolonged PFS compared to those receiving letrozole alone. This combination therapy exemplifies how targeting the cell cycle machinery can enhance therapeutic efficacy.104 This finding is supported by multiple studies, including the PALOMA-2 and PALOMA-3 trials, which demonstrated that palbociclib combined with letrozole or fulvestrant significantly improved median PFS in hormone receptor-positive, HER2-negative advanced breast cancer.105, 106
Furthermore, CPIs such as pembrolizumab and nivolumab, which target PD-1/PD-L1 pathways, have revolutionized cancer therapy by enhancing the immune system’s ability to target tumors. Combining these CPIs with CDK4/6 inhibitors has shown potential in preclinical models to improve antitumor responses by modulating the tumor microenvironment and enhancing immune-mediated tumor cell killing.107
Finally, analyzed studies have demonstrated that various cytogenetic and molecular genetic abnormalities in cancer cells contribute to resistance against standard anti-cancer treatments. The data previously discussed suggest that evaluating the status of the p16 gene and P16INK4a protein offers valuable prognostic information. This assessment can be instrumental in personalizing or targeting therapy plans, thereby improving treatment outcomes.
Limitations
This systematic review faces one main limitation. The key issue is study heterogeneity. The inclusion of studies with differences in population demographics, intervention methods, study designs, and methodological quality presents a challenge to the consistency of the synthesized findings. This variation across the included studies makes it more difficult to combine and interpret the results.
Conclusions
The literature review elucidates the intricate interplay between the physiological functions and altered expression patterns of the p16 gene and its corresponding P16INK4a protein, discernibly linked to the etiology of select hematological malignancies. Noteworthy is the recognition by select authors, whose work is surveyed within the review, of the p16INK4a gene and its protein product as discerning prognostic markers, underscored by their prognostic relevance concerning key clinical endpoints such as OS and EFS.
Furthermore, scholarly discourse extends to deliberations on the potential role of the p16 gene and P16INK4a protein in facilitating the customization and refinement of anticancer therapeutic regimens, thereby accentuating the burgeoning scope for prospective inquiry and scholarly elucidation.
Nevertheless, it is incumbent upon researchers to acknowledge that our current comprehension of the involvement of the p16 gene and P16INK4a protein in the pathogenic cascade of numerous cancers remains only partially elucidated. This caveat is particularly salient within the context of hematological malignancies, a notably diverse and expansive spectrum. Consequently, there exists a manifold of disease entities yet to be scrutinized for putative correlations with the p16 gene and its protein counterpart. Moreover, the presence of incongruences in findings across select investigations underscores the exigency for sustained scholarly engagement and methodological refinement, thereby underscoring the imperative for a meticulous and iterative exploration of this intricate domain to furnish the scientific community with the most cogent and comprehensive insights.