Abstract
Background. The activity of proton pump inhibitors (PPIs) hinders the function of proton pumps that generate stomach acid. Nuclear factor kappa B (NF-κB) is a transcriptional factor engaged in inflammation, immunity and the formation of cancer. The farnesoid X receptor (FXR) is a nuclear receptor that governs the metabolism of bile acids and the metabolic functioning of the liver. The impact of PPIs on the signaling of FXRs and NF-κB is not well understood.
Objectives. We aimed to study the effects of esomeprazole on FXRs and NF-κB signaling in liver cells.
Materials and methods. For the liver cell model, we used the human liver cell line HepaG2. Cells were treated with lipopolysaccharides (LPS) and esomeprazole, and then we assessed the effects of esomeprazole on inflammatory and carcinogenic markers, NF-κB and FXR. We applied the techniques of western blotting, reverse-transcription polymerase chain reaction (RT-PCR), confocal microscopic imaging, and electrophoretic mobility shift assay (EMSA).
Results. Lipopolysaccharides-induced FXRs and NF-κB signaling upregulated the NF-κB-associated cytokines interleukin 6 (IL-6), cyclooxygenase-2 (COX-2) and tumor necrosis factor alpha (TNF-α). Esomeprazole inhibited the upregulation of all these cytokines. Additionally, esomeprazole inhibited LPS-induced FXR expression and NF-κB signaling in HepaG2 cells. The net effect on FXRs and NF-κB signaling was the lower levels of the associated inflammatory and carcinogenic cytokines.
Conclusions. Our study provides insight into the potential therapeutic effects of esomeprazole on hepatic inflammation and carcinogenesis by inhibiting LPS-induced NF-κB and FXR expression in HepG2 cells.
Key words: proton pump inhibitors, nuclear factor kappa B, farnesoid X receptor, lipopolysaccharides, HepaG2
Background
Proton pump inhibitors (PPIs) reduce the amount of acid produced in the stomach by inhibiting the action of proton pumps and are commonly used to treat conditions such as gastroesophageal reflux disease, peptic ulcer disease and Zollinger–Ellison syndrome.1 The most commonly used PPIs include omeprazole, pantoprazole, lansoprazole, and esomeprazole. Clinically, PPIs have side effects such as headache, nausea, diarrhea, renal injury, and an increased risk of bone fractures,2, 3 and the prolonged use of PPI is an important issue. The long-term use of PPIs has been suggested to increase the risk of liver carcinogenesis, including hepatocellular carcinoma (HCC) and cholangiocarcinoma.4, 5, 6 However, evidence is conflicting regarding the association between PPIs and HCC.7, 8
The mechanisms for which PPIs increase the risk of hepatic carcinogenesis are not fully understood, but is worth studying. Several mechanisms could be considered in PPI and hepatic carcinogenesis, including the altered gut microbiota, promoting the growth of harmful bacteria resulting in liver inflammation9, 10, 11 and hypergastrinemia. Gastrin is a hormone that possibly promotes liver cell proliferation, overgrowth and the development of tumors.12 Conversely, several studies have suggested there is no association between PPI use and hypergastrinemia or carcinogenesis.13, 14
Nuclear factor kappa B (NF-κB) signaling is a transcription factor involved in inflammation and carcinogenesis. Nuclear factor kappa B is activated by various stimuli, including inflammatory cytokines, oxidative stresses, and viral or bacterial infections.15, 16 Activation of NF-κB also affects hepatic cancer cells by regulating cell proliferation and apoptosis.17, 18, 19 Thus, NF-κB signaling has been implicated in carcinogenesis,20 and the sustained activation of NF-κB in liver cells could contribute to hepatic carcinogenesis.21 The other nuclear receptor, farnesoid X receptor (FXR), is also expressed on hepatocytes and has bile acid metabolism and inflammation functions. The activation of FXR is recognized for its antitumor properties in the liver, like inhibition of liver tumor cell growth and proliferation, as well as induction of apoptosis in these cells.22
The expression and interaction between FXR and NF-κB in liver carcinogenesis is complex and not well understood. Farnesoid X receptor activation may inhibit NF-κB signaling and reduce hepatobiliary inflammation, thereby preventing tumor development.23
The effects of FXR and NF-κB signaling vary depending on the specific cellular and molecular context. This variability highlights the existing knowledge gap in understanding the mechanisms underlying the effects of PPIs on liver carcinogenesis and the intricate interplay between FXR and NF-κB. Further research in this area is essential to shed light on these complex interactions and their implications for liver health.
Objectives
This study aimed to investigate the effect of esomeprazole on hepatic carcinogenesis by examining the expression of NF-κB and FXR.
Materials and methods
Cell culture
We cultured HepG2 cells, a human HCC cell line, in sterile flasks. The culture medium included Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine and 1% penicillin-streptomycin. For sub-confluent cultures, HepG2 cells were seeded at a density of 2 to 4×104 cells/cm2, while for confluent cultures, we used a density of 8 to 10×104 cells/cm2. Cells were incubated at 37°C in a humidified incubator with 5% CO2, with the culture medium replenished every 2–3 days, or as needed, to maintain optimal cell growth. After reaching 80–90% confluence, cells were passed to a new culture generation. First, cells were rinsed with phosphate-buffered saline (PBS) to remove serum and residual media. Then, trypsin-EDTA (ethylenediaminetetraacetic acid) was used to detach cells from the culture surface. Trypsin was neutralized with fresh media, and the cells were subsequently collected in pellets by centrifugation.
Real-time quantitative polymerase chain reaction
Total RNA was extracted from the cell cultures using the Trizol reagent. Genomic DNA was eliminated with DNase using the Turbo DNA-free kit (Applied Biosystems, Waltham, USA). Next, 1 µg of total RNA was reverse transcribed into Oligo (dT) using the Superscript III First-Strand Synthesis Super Mix (Invitrogen, Waltham, USA). Polymerase chain reaction (PCR) analysis was conducted using the Quant Studio 3 Real-Time PCR System (Thermo Fisher Scientific, Waltham, USA). Two microliters of cDNA were used as the template for PCR amplification, employing interleukin 6 (IL-6) primers (Supplementary Table 1).
Western blot analysis
Cells were first lysed in radioimmunoprecipitation assay (RIPA) buffer (R0278; MilliporeSigma, St. Louis, USA) supplemented with a protease inhibitor. Protein lysates were incubated on ice for 30 min with repeated vortexing every 5 min, and finally cleared by centrifugation at 6,000 g for 15 min at 4°C. Supernatants were then collected and stored at –80°C until further analysis. Protein concentrations in the extracts were determined using the BCA Protein Assay Kit (23225; Thermo Fisher Scientific). Aliquots of 30 μg of total protein extracted from cultured cells were subjected to electrophoresis on a 10% sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) at a constant voltage of 70 V for 30 min, followed by 110 V for 90 min in a running buffer (25 μM Tris-HCl pH 8.3, 192 μM glycine and 0.1% SDS).
Subsequently, proteins were transferred from the gel to a nitrocellulose membrane using a constant current of 350 mA for 90 min in a transfer buffer (25 μM Tris-HCl pH 8.3, 150 μM glycine and 5% v/v methanol). The nitrocellulose membrane was then blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS; 20 μM Tris pH 7.6, 137 μM NaCl) for 1 h. Immunostaining was performed using a monoclonal FXR (NR1H4) antibody and Phospho-NF-κB p65 (Ser536) antibody (both at 1/1,000 dilution; Cell Signaling Technology (CST), Danvers, USA), followed by incubation with a secondary polyclonal mouse anti-goat antibody conjugated to horseradish peroxidase (HRP) (diluted at 1!/5,000; CST). Blots were developed using HRP and Trident Femto Western HRP Substrate (GTX14698; Gene Tex, Irvine, USA). Data acquisition was performed using a Geliance CCD camera (PerkinElmer, Waltham, USA). Image analysis was carried out using ImageJ software (National Institutes of Health (NIH), Bethesda, USA)
Confocal microscopy analysis
Laser confocal microscopy (Olympus FV1000; Olympus Corp., Tokyo, Japan) was employed to localize subcellular proteins, including NF-κB. Immunofluorescence staining was performed to label samples with a fluorescently conjugated NF-κB specific antibody. Subsequently, confocal microscopy was conducted using a confocal laser scanning microscope.
Sample preparations involved the following steps. First, cells were fixed and permeabilized, and the nonspecific binding sites were blocked using a blocking agent (e.g., BSA or serum). Next, samples were incubated with a primary antibody specific to NF-κB while diluted in a blocking buffer. Afterward, samples were washed to remove any unbound primary antibodies. Samples were then incubated with a secondary antibody conjugated to a fluorescent dye, diluted in a blocking buffer, and washed again to eliminate any unbound secondary antibodies. Finally, samples were mounted on a microscope slide using a mounting medium, and images were taken with a confocal laser scanning microscope.
Electrophoretic mobility shift assay
HepG2 cells were first treated with esomeprazole and lipopolysaccharide (LPS), before being harvested to obtain nuclear extracts according to the standard procedures as described in the literature. To perform electrophoretic mobility shift assay (EMSA), we used a double-stranded DNA probe containing the consensus sequence for the NF-κB binding site (5’-GGGACTTTCC-3’), which was labeled with biotin. The labeled DNA probe was incubated with the nuclear extract in a binding buffer containing nonspecific competitor DNA (such as poly dI-dC) and a specific competitor DNA containing the NF-κB binding site. The specific competitor DNA was obtained commercially or prepared by annealing oligonucleotides containing the NF-κB binding site.
The binding reaction was conducted at room temperature for 30 min to allow the formation of NF-κB-DNA complexes. Subsequently, the reaction mixture was loaded onto a non-denaturing polyacrylamide gel, which allowed electrophoresis to separate the NF-κB-DNA complexes from the free DNA probes. After electrophoresis, gel visualization was achieved using autoradiography for the radioactive probes. As the NF-κB DNA complex migrated more slowly than the free DNA probe, a shifted band appeared on the gel, indicating the presence of the complex.
Statistical analyses
Each group in this study was examined in each experiment. Results were presented as medians. The quantitative polymerase chain reaction (qPCR) results shown in the dot plots were obtained from 6 independent experiments (n = 6). All experiments were independently repeated 6 times. The Kruskal–Wallis test was conducted, and if differences were significant, the Dunn–Bonferroni test was applied. Differences were considered statistically significant at a p-value < 0.05. For data analysis, we used IBM SPSS v. 22.0 (IBM Corp., Armonk, USA).
Results
Lipopolysaccharides increased the expression of NF-κB signaling molecules in HepG2 cells
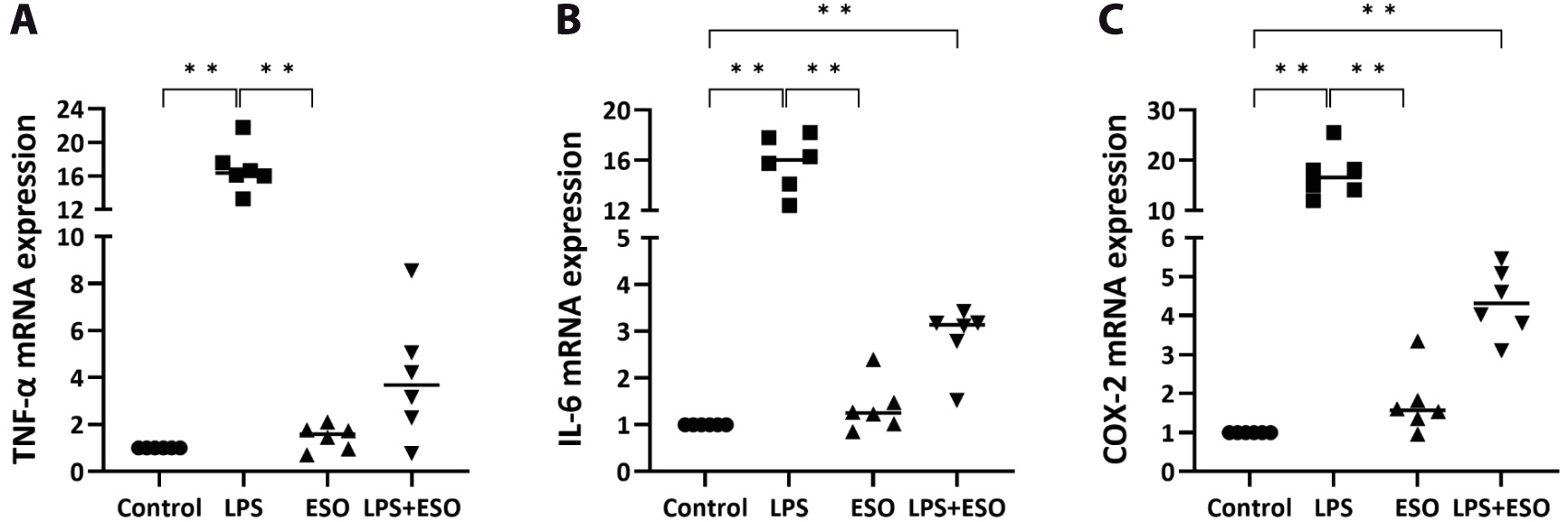
Lipopolysaccharides, a component of the outer membrane of Gram-negative bacteria, activates various signaling molecules in host cells, including the NF-κB transcription factor. Such LPS activation plays a crucial role in regulating the expression of genes related to inflammation, immunity and cell survival. We found that LPS treatment increased NF-κB expression and activated downstream targets by increasing the expression of inflammatory cytokines, such as IL-6, cyclooxygenase-2 (COX-2) and tumor necrosis factor alpha (TNF-α) (Figure 1).
Lipopolysaccharides increased FXR expression in HepG2 cells
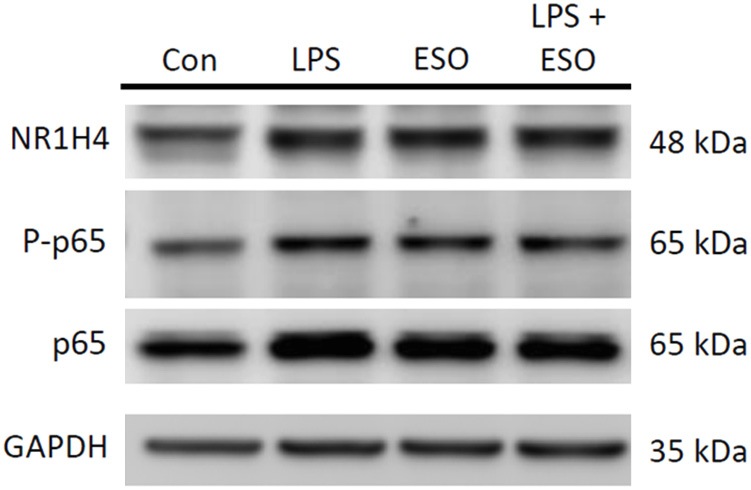
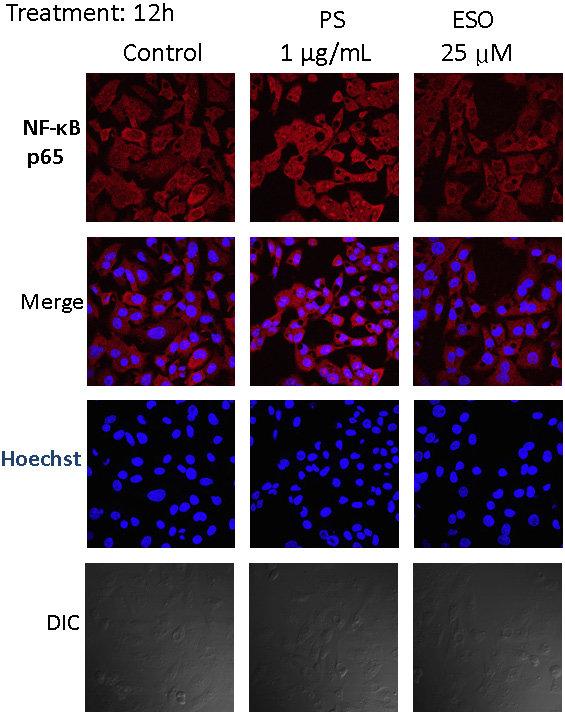
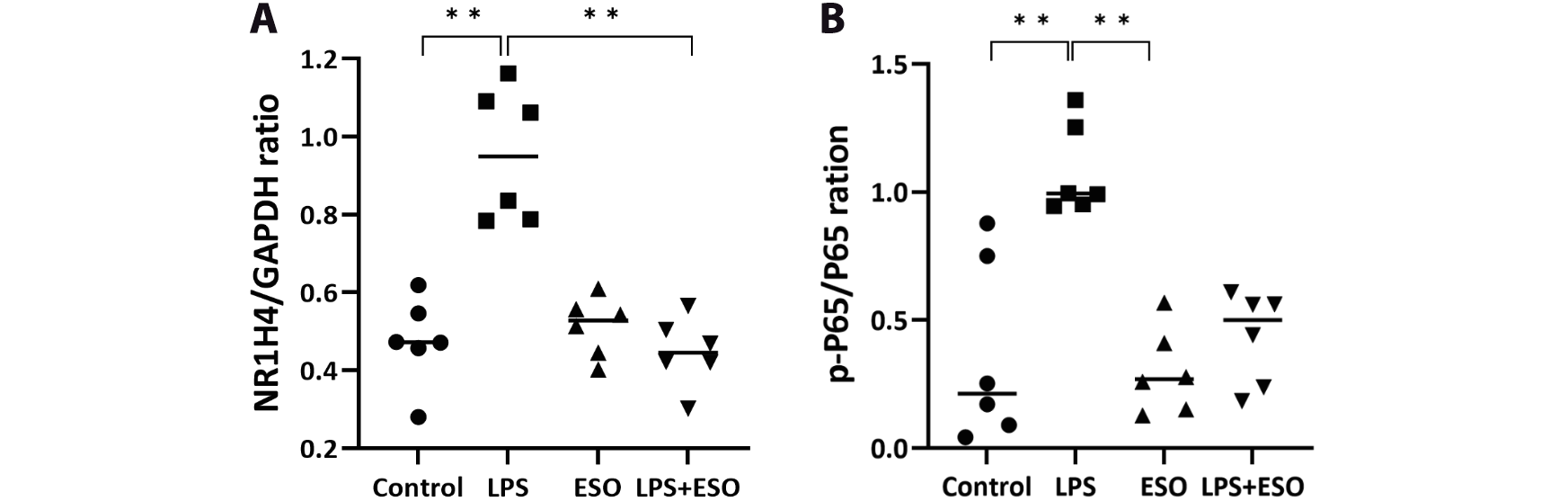
Lipopolysaccharides are known to increase FXR expression in various cells, including hepatocytes and macrophages. Activation of the FXR by LPS has been proposed to play a protective role in various diseases by regulating the expression of genes involved in inflammatory and immune responses. Figure 2 shows that LPS treatment for 12 h increased FXR expression in HepG2 cells. The confocal microscopic images in Figure 3 demonstrate that esomeprazole inhibited LPS-induced NF-κB expression. Based on these results, esomeprazole, in addition to treating acid-related disorders, likely exerts an anti-inflammatory effect by blocking the LPS activation of NF-κB. Figure 4A illustrates that LPS treatment significantly upregulated the expression of FXR in HepG2 cells compared to the untreated control group. This upregulation was significantly mitigated when the cells were co-treated with esomeprazole, indicating that esomeprazole effectively downregulated LPS-induced FXR expression (Supplementary Table 2).
Esomeprazole inhibited the expression of NF-κB and FXR in HepG2 cells
Furthermore, as shown in Figure 4B, LPS treatment resulted in a significant elevation in p-P65/P65 expression, highlighting an increase in NF-κB activation. Co-treatment with esomeprazole led to a reduction in the LPS-induced p-P65 expression, although this reduction was borderline significant. The statistical significance of these findings was assessed using the Kruskal–Wallis test, which indicated significant differences among the treatment groups. Subsequent post hoc comparisons were conducted using Dunn–Bonferroni tests. Detailed p-values from both the Kruskal–Wallis and Dunn–Bonferroni tests are provided in Supplementary Table 2.
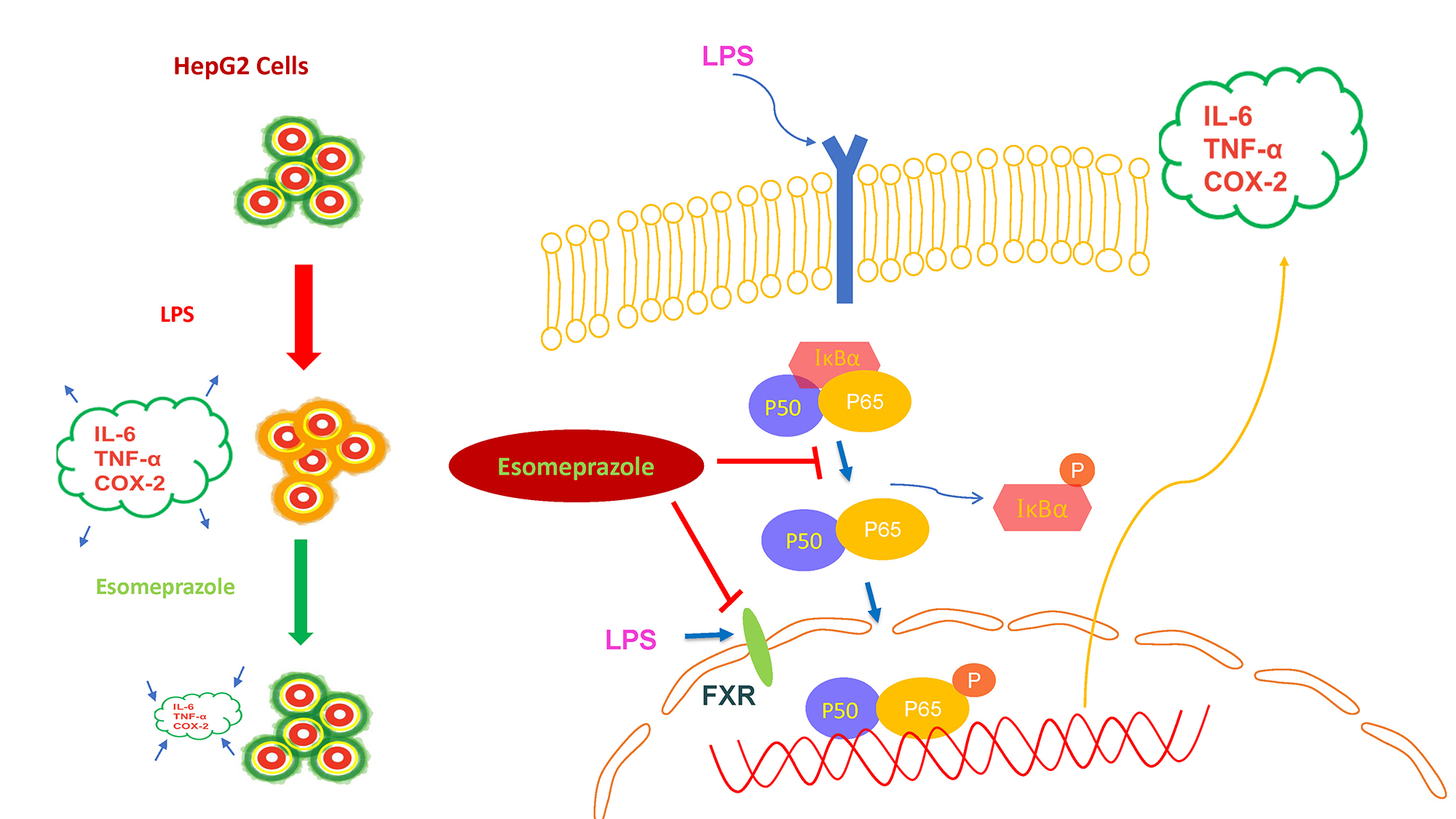
These results suggest that esomeprazole has a modulatory effect on LPS-induced changes in NF-κB and FXR signaling pathways in HepG2 cells. Specifically, esomeprazole appears to counteract the pro-inflammatory effects induced by LPS, potentially through the regulation of the FXR and NF-κB signaling.
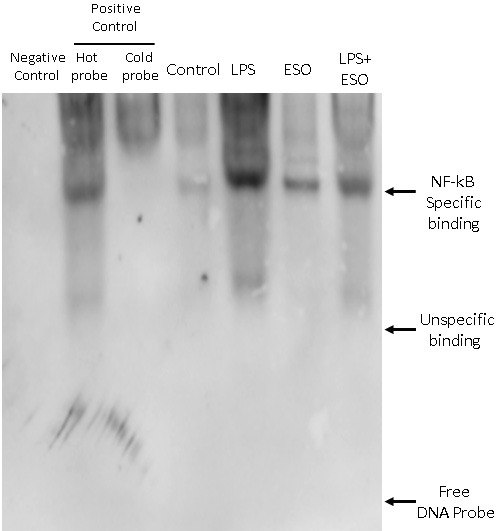
Electrophoretic mobility shift assay showed esomeprazole inhibition of LPS-induced NF-κB B expression
To investigate whether esomeprazole inhibits LPS-induced NF-κB expression, we conducted an EMSA by incubating a radiolabeled DNA probe containing an NF-κB binding site with nuclear extracts from LPS-treated cells, in the presence and absence of esomeprazole. If esomeprazole inhibits NF-κB expression in the presence of esomeprazole, EMSA should have revealed a diminished band pattern corresponding to the NF-κB-DNA complex. That was exactly what we observed (Figure 5).
Discussion
Esomeprazole, a PPI, is widely used to reduce acid production in the stomach to treat acid-related disorders and has proven clinical outcomes. Thus, the long-term use of PPIs has been considered an important issue in medical care. Nuclear factor-kappa B is a transcription factor involved in the regulation of inflammation and carcinogenesis. In the present study on HepaG2 cells, we found that esomeprazole inhibited LPS-induced FXR and NF-κB expression.
An interaction between FXRs and NF-κB has been observed in the liver. The FXR activation inhibits NF-κB activation, resulting in the downstream production of pro-inflammatory cytokines that contribute to liver injury and inflammation. On the other hand, NF-κB activation promotes liver inflammation and injury by promoting the production of pro-inflammatory cytokines, such as TNF-α and IL-6, leading to the progression of liver disease. Therefore, targeting FXRs and NF-κB is likely a complementary approach to treating liver diseases related to inflammation and injury. In our study, we found both FXRs and NF-κB are activated by esomeprazole in LPS-treated hepatocytes. The inflammatory as well as carcinogenesis effects were both inhibited by the PPI in HepG2 cells.
Proton pump inhibitors may play a protective role in inflammation and carcinogenesis. In studies of nerve cells, PPIs exert mitochondrial apoptosis and attenuate NF-κB signaling, suggesting its potential as an effective and safe anticancer treatment for gliomas.24 An animal study found that PPIs inhibit the induction of several inflammatory mediators, including cytokines, chemokines and nitric oxide (NO), by suppressing NF-κB, thereby preventing fulminant liver failure.25
The PPI effect on the process of carcinogenesis may be paradoxical. Proton pump inhibitors paradoxically augment the anti-tumorigenic and gastrin in an APCMin/+ intestinal polyposis animal model.26 Our study further demonstrated the potential inhibitory effect of esomeprazole on NF-κB and its associated signaling pathways in hepatocytes.
The interaction between FXR and NF-κB in the liver is complex and not fully understood. Wang et al. demonstrated that the FXR acts critically as a negative mediator of hepatic inflammation, contributing to hepatoprotection and suppressing hepatic carcinogenesis.23 However, in our study, we found that both FXR and NF-κB signaling were inhibited by esomeprazole in LPS-induced hepatic inflammation. The net effect of the inhibition of the FXR and NF-κB is reduced levels of associated cytokines.
Limitations
The study primarily uses HepaG2 cells, which is a common cell line for liver-related research. However, the relevance of findings in cell lines to clinical outcomes in humans is often limited and may not fully represent the complexity of liver-related diseases. This article discusses the potential benefits of targeting FXRs and NF-κB in liver diseases, but it may not fully capture the diverse nature of liver-related disorders, which can have multiple underlying causes and mechanisms.
Conclusions
Our study provides insight into the potential therapeutic effects of esomeprazole on hepatic inflammation and carcinogenesis by inhibiting LPS-induced NF-κB and FXR expressions in HepG2 cells. However, more research is needed to fully understand its effects on gene expression and cellular pathways in various contexts, including in vivo and human studies.
Supplementary data
The Supplementary materials are available at https://doi.org/10.5281/zenodo.12740310. The package includes the following files:
Supplementary Table 1. Primers sequence used for RT-PCR.
Supplementary Table 2. HepG2 cells were treated with or without lipopolysaccharides or esomeprazole, and compared to untreated cells using qPCR.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.