Abstract
Neurodegenerative diseases, including Alzheimer’s and Parkinson’s disease, affect an increasing number of people in aging societies, dramatically reducing the quality of life of those affected. Hence, intensive research efforts are aimed at understanding the molecular mechanisms of the disease progress, with the hope for developing effective therapeutic strategies. The progress of neurodegenerative diseases is associated with a complex activity of the immune system in the brain tissue. Carbohydrate-binding proteins (lectins) play a key role in the inflammation-related activation of microglia. Siglecs, maintained in an active state by binding to sialic acid-terminated glycoconjugates, help establish homeostasis by protecting nerve cells from phagocytosis and preventing triggering receptor expressed on myeloid cells 2 (TREM2) activation. Upon activation, microglia release sialidase, an enzyme that cleaves sialic acid residues from glycoconjugates, thereby exposing galactose as the next monosaccharide in the glycan chain. After losing siglec-mediated protection, the glycan becomes a ligand for Galectin-3 (Gal-3). Overexpression of this lectin under inflammatory conditions activates TREM2 and TLR4 signaling pathways, enhances the phagocytic activity of microglia and leads to tissue damage. Blocking Gal-3 interactions with the thiodigalactoside inhibitor (TD-139) appears to be a promising novel approach to pharmacologically alleviating neuroinflammation.
Key words: microglia, neurodegeneration, galectin, glycosylation, Siglec
Introduction
Aging populations are experiencing a rising incidence of neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease. The progressive degradation of the nervous tissue significantly reduces the quality of patient’s life, inevitably leading to disability. The scale of the problem becomes a serious challenge for the healthcare systems. Despite extensive research, the precise mechanisms driving the onset and progression of these diseases remain poorly understood, and the search for regulatory targets to support the development of novel therapeutic strategies is ongoing.
Activation of the immune system is one of the mechanisms involved in the development of neurodegenerative diseases. When immune cells are unable to effectively remove deposits of misfolded proteins, they intensify their activity, which eventually leads to an attack on the body’s own nervous tissue. Protein–carbohydrate interactions are important in regulation of these processes, and carbohydrate-recognizing proteins – lectins – act as pattern recognition receptors. In the recent years, attention has been paid to the interactions mediated by galectins, particularly Galectin-3 (Gal-3).1, 2, 3, 4, 5 Galectins are a group of lectics, capable of recognizing β-galactosides in the oligosaccharides of glycoconjugates. Protein–sugar interactions mediated by lectins are involved in numerous physiological and pathological processes, including those related to cancer, infections and autoimmune diseases.6 The key issue seems to be the regulation of the immune response, for which the subtle changes in the sugar structures of glycoproteins and glycolipids are essential.
A significant part of the brain tissue is made up of the immune system cells, mainly astrocytes and microglia. As in other tissues, they act as guardians, detecting compounds considered non-self in their surroundings.7, 8, 9 In nervous tissue, the inflammatory process is most often independent of pathogens. The immune response can be triggered by tissue damage or the accumulation of misfolded protein deposits, such as β-amyloid in AD or fibrillar forms of α-synuclein in Parkinson’s disease.4 Such a trigger leads to the activation of microglia (Figure 1). The resting ramified microglial cells change their shape, transcriptional profile and behavior, transforming into an amoeboid form capable of phagocytosis.7 Such an inflammatory state is of the key importance in the pathology of neurodegenerative diseases. With the initial appearance of deposits of misfolded, insoluble protein plaques, the activation of microglial cells is aimed at the clearance of debris and thus maintaining a healthy tissue profile.6, 7, 8 Later, the active microglia secrete significant amounts of pro-inflammatory cytokines, and the increasing inflammation leads to the progressive damage of the nervous tissue. An altered transcriptional profile of activated microglia leads to the massive release of cytokines and other inflammation-associated proteins, including Gal-3. This protein is weakly expressed in homeostatic, inactive microglia, but is highly overexpressed in activated cells (Figure 1).2, 3, 8, 9, 10
Glycosylation as an immune checkpoint
Glycosylation is a common post-translational modification of proteins. Oligosaccharide chains decorate the surface of more than half of the proteins in all living organisms. Membrane lipids, such as gangliosides, are also glycosylated and are particularly abundant in nervous tissue. Even subtle changes in structures of these oligosaccharides have essential impact on the dynamics of interactions with their receptors.11
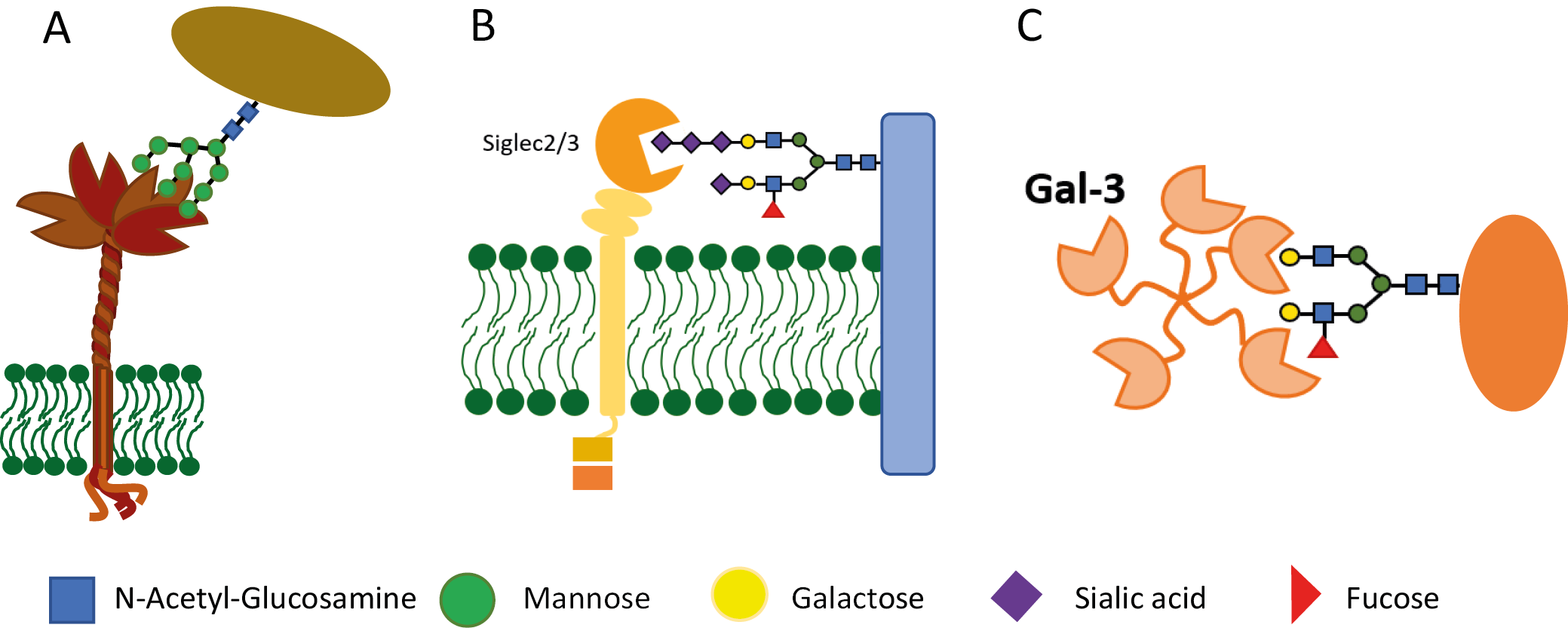
Complex glycosylation pathways enable the creation of countless variants of oligosaccharide structures. As a result of evolution, these structures differ between lower organisms (including pathogens) and vertebrates. In vertebrates, tissue-specific and developmental (ontogenetic) differences also play a significant role. Structural changes in glycans are associated with numerous pathological processes, including carcinogenesis. The diverse variants of oligosaccharides are recognized by an equally rich array of proteins, collectively known as lectins (Figure 2).11 In addition to the previously mentioned galectins, there are groups of lectins that recognize high-mannose glycans and are responsible for detecting and binding pathogens. The 3rd major group comprises Siglecs, which specifically recognize sialic acid-containing structures. Lectins are classified as pattern recognition receptors (PRRs), with the “pattern” referring to specific glycosylation profiles.12 Pattern recognition drives the appropriate immune response of the organism. Thus, mannose-specific lectins primarily recognize pathogen-associated molecular patterns (PAMPs).13, 14 In the context of the topics discussed here, 2 additional patterns are of particular interest: damage-associated molecular patterns (DAMPs), recognized by galectins, and self-associated molecular patterns (SAMPs), recognized by Siglecs, i.e., the sialic acid-binding immunoglobulin-like lectins. Glycosylated ligands for these lectins can be either anchored to the cell membrane or secreted into the extracellular space (Figure 2).4, 15, 16, 17
Sialylation, Siglecs and sialidase
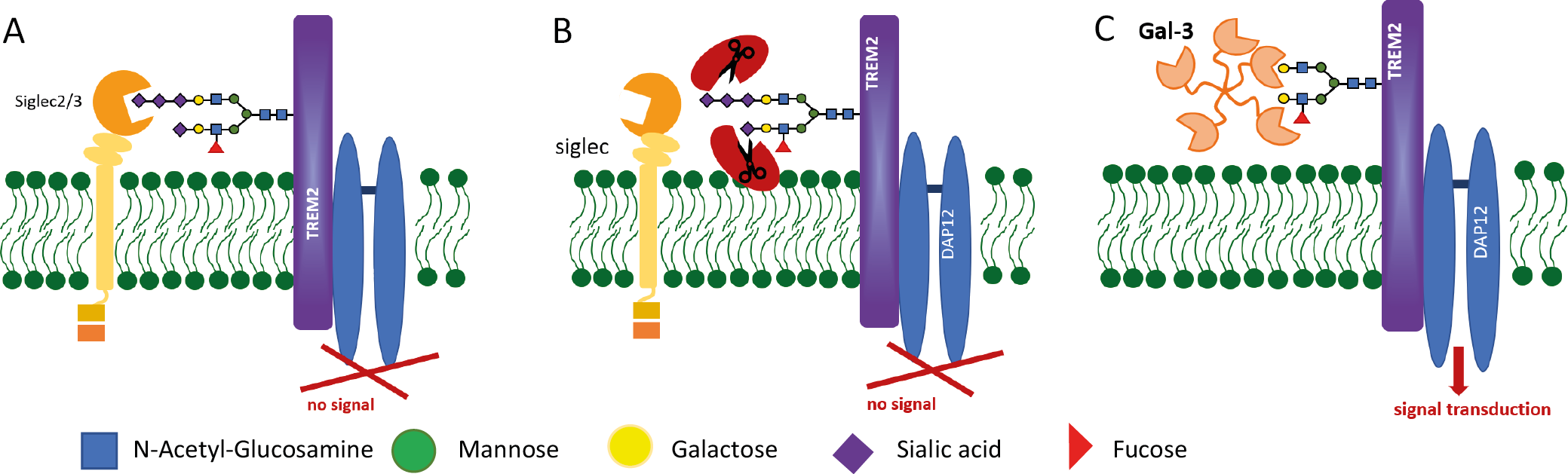
The vast majority of glycoproteins and glycolipids in the human body are terminated with a sialic acid residue, a monosaccharide that consistently occupies the terminal position within oligosaccharide structures, although the mode of attachment can vary.11 This characteristic also applies to nervous tissue. Moreover, particularly in the central nervous system (CNS), glycans often form unique structures characterized by the presence of multiple sialic acid residues at their termini (polysialylation).18, 19 Their receptors, the Siglecs, are expressed on the surface of both neurons and microglial cells.17, 18, 20 This protein–carbohydrate interaction is responsible for maintaining homeostasis. Activation of CD22 (Siglec-2) and CD33 (Siglec-3) through binding to sialylated ligands inhibits phagocytosis, suppresses TLR4 activation and restricts complement receptor 3 (CR3) access to the cell surface.18, 19, 21 The situation changes when a trigger signal emerges within the tissue, leading to microglial activation. The initial deposits of amyloid-β or other misfolded proteins may serve as such a trigger.22 In response, microglial cells secrete sialidase (also known as neuraminidase), an enzyme that specifically cleaves terminal sialic acid residues from the surface glycans of both microglia and neurons.16, 19, 21, 23, 24 The removal of sialic acid exposes the underlying galactose residue within the glycan chain, thereby disrupting the interaction with Siglecs that normally serves to protect cell integrity. Moreover, nerve fibers with exposed galactose residues become susceptible to phagocytosis and, additionally, serve as ligands for Gal-3.19 Thus, in resting microglia, the interaction between Siglecs and their ligands is crucial for maintaining cellular homeostasis. Upon activation, sialidase-mediated modification of glycan structures disrupts this protective mechanism.
Galectins in the nervous tissue
Microglial activation is primarily directed toward the clearance of early deposits of misfolded proteins.3, 7 At this stage, the cells start to express and secrete Gal-3. Galectin-3, released from activated microglia, further amplifies microglial activation by binding to TLR4 and TREM2, attaches to desialylated neurons to opsonize them for phagocytosis, and promotes amyloid-β aggregation and associated toxicity (Figure 3).1, 2, 3, 4, 25, 26, 27
Galectin-3, the most extensively studied galectin in CNS pathology, is the sole representative of the chimeric type within this protein family (Figure 3). In addition to its C-terminal carbohydrate recognition domain (CRD), Gal-3 possesses a flexible collagen-like domain and an N-terminal domain responsible for multiple protein–protein interactions.3, 4, 10, 28 This N-terminal domain facilitates oligomerization, allowing Gal-3 to form higher-order structures, including pentamers. Due to its unique structure, Gal-3 can therefore interact with both sugars and protein structures, which is an exception in this type of lectins. Multimeric CRD interactions, as well as simultaneous interactions of the CRD and N-terminal domains, give Gal-3 the ability to function as a bridge, connecting ligands on the cell surface, in the extracellular matrix (ECM), and elements secreted outside the cell with those anchored in the membrane. Additionally, Gal-3 is susceptible to 2 characteristic post-translational modifications: proteolytic cleavage by matrix metalloproteinases (MMPs), separating the CRD domain from the N-terminal fragment, and phosphorylation. N-terminal cleavage prevents oligomerization and simultaneous interactions with both carbohydrate and protein ligands. The role of potential phosphorylation remains unknown.
Galectin-3 levels in cerebrospinal fluid (CSF) and serum of AD patients are significantly elevated and correlate with disease stage and severity. A similar increase was observed in mouse models of AD, particularly in microglial regions associated with amyloid deposits.19, 29, 30 Studies have demonstrated that the loss of Gal-3 function, via genetic deletion or inhibition, mitigates key features of neurodegenerative diseases, including decreased expression of pro-inflammatory genes, reduced amyloid burden and improved cognitive performance.10, 29 In the healthy CNS, the role of Gal-3 seems to be limited. Knockout mice (Gal-3–/–) do not exhibit any significant functional impairments.3, 28 However, in healthy neural tissue, Gal-3 is physiologically expressed in the subventricular zone, where neurogenic stem cells reside. Microglia in this area are semi-activated despite the absence of tissue damage. This process is reported to be associated with neurogenesis and migration of new neurons to olfactory bulbs.10
Apart from Gal-3, expression of Galectin-1, -3, -8, and -9 has been described in the nervous tissue.31, 32, 33, 34, 35 Galectins other than Gal-3 are considered neuroprotective rather than promoting a tissue damage. Galectin-1 belongs to the prototype group of galectins. These proteins contain a single CRD capable of dimerization, enabling bivalent interactions and cross-linking of glycosylated ligands. Galectin-1 is often co-expressed with Gal-3 but exerts opposing effects. It is highly expressed in astrocytes and plays a regulatory role by suppressing microglial activation. This is achieved through binding to O-glycans on CD45 and attenuating inflammatory signaling pathways. In Gal-1 knockout mice, microglia display a more pro-inflammatory phenotype, accompanied by exacerbated demyelination.36 Galectins-4, -8 and -9 belong to the tandem-repeat type, characterized by the presence of 2 distinct CRDs connected by a polypeptide linker of variable length. This structure also allows ligands cross-linking. Galectin-8, similarly to Gal-9, has an immunosuppressive impact on the CNS. It is involved in the activation of autophagy in cells containing tau aggregates in AD. Galectin-8 detects luminal glycans of the damaged endomembranes caused by cytosolic aggregation of tau protein and directs them to autophagy. Inhibition of Gal-8 results in the higher amount of tau aggregates inside the cells.34 Galectin-4 is localized in neurons and plays its role in axonal growth and transport. Its presence is also associated with the inhibition of myelination and the maturation of oligodendrocytes. However, no significant changes have been reported in the overall progression of neurodegeneration.36
Cross-talk of Siglecs, Galectin-3 and TREM2
Over the past decade, triggering receptor expressed on myeloid cells 2 (TREM2) has been considered implicated in the progression of AD.37, 38, 39 Expressed by microglia, TREM2 is a key regulator of the innate immune response. It is a membrane protein composed of a large ectodomain that binds extracellular ligands, a transmembrane region consisting of a single α-helix, and a short cytoplasmic tail lacking intrinsic signaling motifs. TREM2 requires interaction with the adaptor proteins DAP12 or DAP10 to activate signaling pathways that promote the secretion of pro-inflammatory cytokines and enhance phagocytic activity.39, 40 Genetic variants of TREM2 have been associated with an increased risk of AD and other neurodegenerative disorders.39 In the CNS, TREM2 expression increases with age and the progression of neurodegenerative diseases.39 Studies using TREM2–/– mice have shown a decrease in microglial clustering around amyloid deposits, reduced microglial cell numbers and increased apoptosis.41 The TREM2 ectodomain can be shed by ADAM-family metalloproteinases, generating a soluble form (sTREM2) that acts as a mediator of cellular interactions within the tissue (Figure 4).42, 43
Both Gal-3 and Siglec-3 (CD33) are endogenous ligands of the TREM2/DAP12 receptor complex, modulating its signaling, a key driver of microglial activation and neurodegeneration, which plays a crucial role in regulating phagocytosis and inflammation.29, 41, 44, 45 The binding of Siglec-3 with sialylated glycans suppresses TREM2-driven phagocytic activity in microglia (Figure 4).41, 44, 46 In turn, increased Gal-3 expression leads to the activation of TREM2 signaling pathways.29, 45 Notably, Gal-3 binding to TREM2 has been shown to involve its carbohydrate recognition domain.29
Therapeutic approach
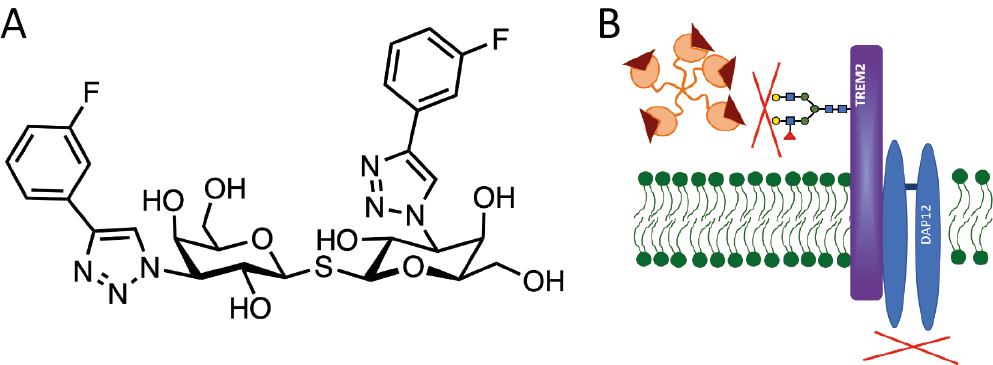
The interaction between terminal sialic acids and Siglecs appears to play a key role in protecting brain tissue, suggesting that inhibiting sialidase may hold therapeutic potential.47 However, current research is focused on low-molecular-weight inhibitors of the galectin CRD. Among a few digalactosyl analogues of lectin natural ligands,48 TD-139 (olitygaltin) is the subject of most advanced studies. The compound, 3,3′-bis-(4-aryltriazol-1-yl)thio-digalactoside, consists of a core formed by 2 galactose units linked via a thiol bond, flanked by triazole rings and fluorine-substituted phenyl groups (Figure 5). The reduction of pro-inflammatory Gal-3 activity by TD-139 has been tested in several animal models of nervous system diseases. Rombaut et al.49 demonstrated a neuroprotective effect of intravitreally injected TD-139 in a rat ocular hypertensive model of glaucoma, reporting the prevention of retinal ganglion cell degeneration. In a mouse model of an early brain injury due to subarachnoid hemorrhage, Shen et al.50 demonstrated both the role of Gal-3 in the microglia activation and its efficient inhibition by TD-139, which prevented brain injury and excessive secretion of pro-inflammatory cytokines by the microglial cells. In a rat model of epilepsy, elevated Gal-3 activity was observed, and its inhibition by TD-139 was shown to reduce inflammation and associated neurodegenerative changes. This intervention reduced the expression of pro-inflammatory factors, the severity of seizures and hippocampal damage.51 Experimental studies in animal models may be limited by the significantly lower binding affinity of the inhibitor to mouse and rat analogues of Gal-3 compared to the human form of the lectin.52 Based on crystallographic studies, the authors indicate structural differences in the carbohydrate-binding site in all 3 cases. The key role is suggested to hA146, which holds the ligand in the correct position in the lectin binding pocket by interacting with the fluorophenyl flanking structure. The presence of larger V160 and T158 residues in mouse and rat proteins disrupted the alignment required for proper binding. Interestingly, targeted mutations replacing the aforementioned residues with alanine in animal proteins led to improved alignment and increased binding affinity. Conversely, introducing a valine residue in place of alanine in human Gal-3 diminished its binding affinity. Such data are essential not only for further planning of animal model studies and their interpretation, but also for possible research on the advantageous structural modifications of digalactosyl compounds (Figure 5).
Conclusions
Interactions between lectins and their carbohydrate ligands are gaining increasing attention as effective immune checkpoints that regulate the body’s immune response. This applies to various diseases that contain an inflammatory component. It does not differ in disorders affecting the nervous tissue, although our knowledge here is much more limited. Further studies are important for a better understanding of the dynamics and regulation of neuroinflammation that leads to a tissue damage. Current data suggest that a deeper understanding of these interactions could pave the way for the development of new therapeutic agents aimed at slowing the progression of neurodegenerative diseases. The challenge for our understanding of these mechanisms comes from the possible pleiotropic effects of lectins, as their action may be context-dependent and vary in changing conditions of the microenvironment. Little is known about the possible interplay of different galectins, sometimes co-expressed but varying in function. The signaling pathways leading to galectin expression may be also influenced by external factors.
Current efforts to utilize the TD-139 inhibitor as a potential therapeutic agent originate primarily from its demonstrated efficacy in treating pulmonary fibrosis by limiting the Gal-3-induced excessive immune response. However, in the context of neurological diseases, an added challenge lies in the limited understanding of its ability to cross the blood–brain barrier. The molecule, in addition to the digalactoside structure recognized by the lectin, contains flanking regions composed of phenyl-derived moieties. Due to its relatively hydrophobic character, TD-139 may more easily penetrate biological membranes, including potentially the blood–brain barrier. This highlights the critical role of the flanking structures surrounding the galactoside core, which may serve as key targets for future modifications aimed at enhancing membrane permeability. Another advantage of low molecular weight compounds like TD-139 lies in their ability to circumvent potential side effects commonly associated with antibody-based therapies, such as immune system activation.
Although our understanding of the complex mechanisms underlying neuroinflammation and degeneration is still incomplete, the topic continues to garner significant research attention, and a surge of new, insightful studies is anticipated in the near future.