Abstract
The U.S. Food and Drug Administration (FDA) recently approved a blood-based biomarker to confirm diagnosis of Alzheimer’s disease (AD-BBMs). When used in conjunction with human expertise in the diagnosis of neurocognitive disorder due to Alzheimer’s disease (AD), blood-based biomarkers could increase both the accessibility and sustainability of medical practice and research. Indeed, AD-BBMs are likely to be more cost-effective in the long term and conservative calculations performed here suggest that they would have an approx. 10-fold and 36-fold lower carbon footprint compared to cerebrospinal fluid (CSF) lumbar punctures and amyloid positron emission tomography (PET) scans, respectively. Their use will require a careful balance of trade-offs to maximize benefits and minimize harms for current patients, while ensuring the sustainable integration of these tools into healthcare systems so that diagnostic precision remains accessible to present and future generations in an aging global population amid anthropogenic climate change.
Key words: biomarkers, Alzheimer’s disease, access, sustainability, climate change
Introduction: A newly-approved blood test to help in the diagnosis of Alzheimer’s disease
According to the World Health Organization (WHO), in 2021, over 55 million people were living with dementia, a clinical syndrome of neurocognitive disorder defined by the coexistence of acquired cognitive impairment, associated with behavioral and psychological symptoms leading to functional impairment.1
Alzheimer’s disease (AD), first described as a rare condition in the early 1900s, became widely recognized in the 1970s as the predominant cause of late-life dementia based on converging clinical and pathological evidence.2 It is now estimated to account for approx. 40 million cases of dementia worldwide. Alzheimer’s disease is a progressive neurodegenerative disorder characterized by the accumulation of amyloid-β and tau proteins in the brain, leading to progressive memory loss, cognitive and functional decline, and neuropsychiatric symptoms.
Historically, AD was defined and confirmed by post-mortem neuropathological examination in people who died with neurocognitive dementia. This neuropathological examination typically revealed the presence of senile plaques containing amyloid-beta outside neurons, and neurofibrillary degeneration containing tau protein inside neurons. For as long as in vivo biomarkers of amyloid and tau protein were not available, in vivo diagnosis of AD was probabilistic. However, since the late 2000s, in vivo biomarkers – positron emission tomography (PET) imaging and cerebrospinal fluid (CSF) lumbar punctures – have been used to confirm diagnosis of AD, mostly in high-income countries.3 These biomarkers are also increasingly used to define, explain, and treat AD.2 Some anti-amyloid antibodies with modest effects on the slowing of cognitive decline in AD have recently been approved in different health systems.4
Against this backdrop, on May 16th 2025, the U.S. Food and Drug Administration (FDA) approved the first blood test for in vitro diagnosis of AD via the FDA Breakthrough Device Designation mechanism:
The Lumipulse G pTau217/β-Amyloid 1-42 Plasma Ratio is for the early detection of amyloid plaques associated with Alzheimer’s disease in adult patients, aged 55 years and older, exhibiting signs and symptoms of the disease … [in] a multi-center clinical study of 499 individual plasma samples from adults who were cognitively impaired … 91.7% of individuals with … Plasma Ratio positive results had the presence of amyloid plaques by PET scan or CSF test result, and 97.3 % of individuals with negative results had a negative amyloid PET scan or CSF test result. Less than 20% of the 499 patients tested received an indeterminate … Plasma Ratio result … the new blood test can reliably predict the presence or absence of amyloid pathology associated with Alzheimer’s disease at the time of the test in patients who are cognitively impaired. The test is intended for patients presenting at a specialized care setting with signs and symptoms of cognitive decline. The results must be interpreted in conjunction with other patient clinical information.5
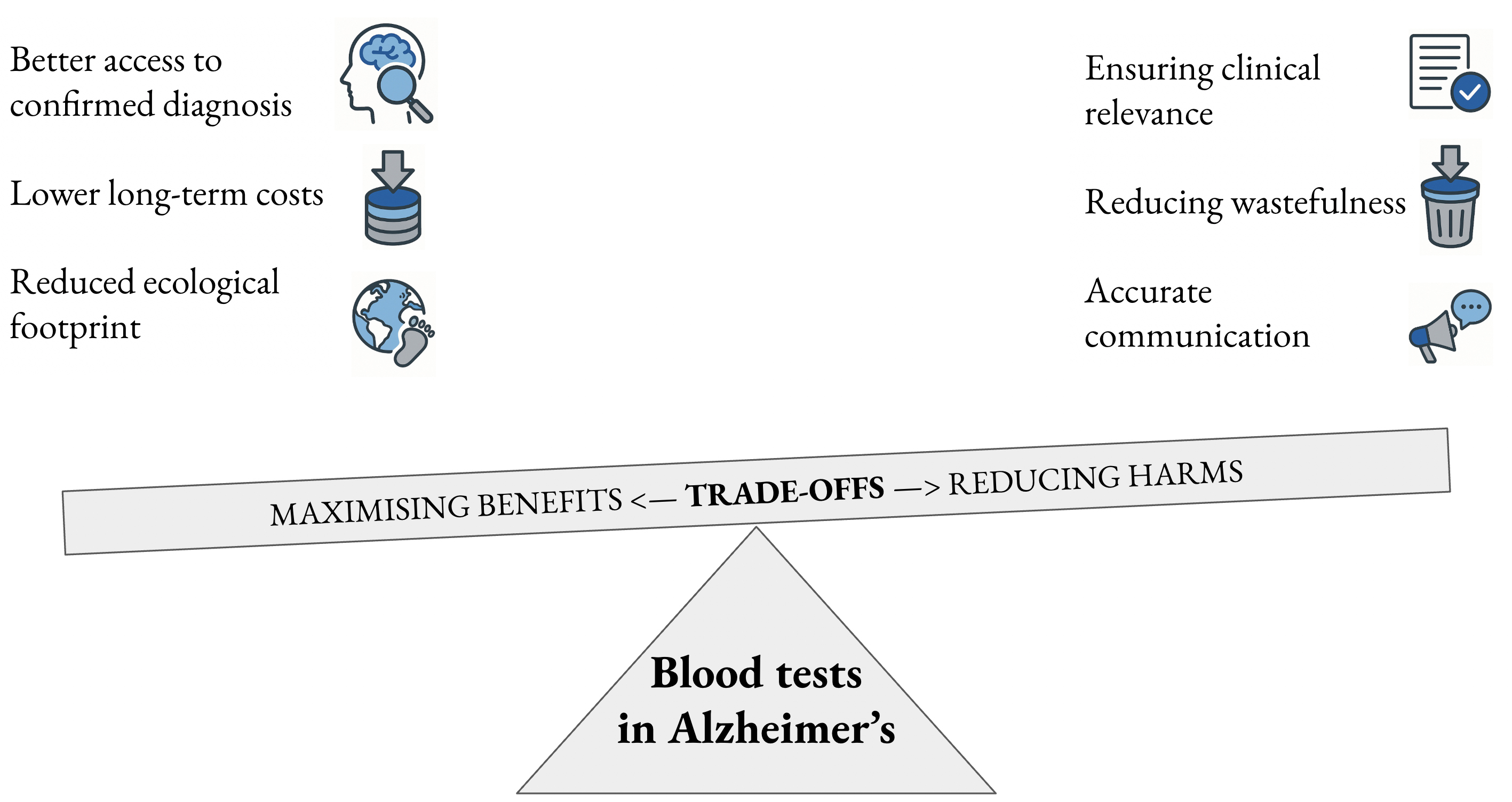
I will analyze this regulatory decision and argue that for the global community to capitalize on this significant moment in the history of AD, there will be an important trade-off to be made between clinical relevance in medical practice, increased accessibility to a diagnosis of AD and improved environmental sustainability of healthcare practices.
In clinical practice, blood tests in AD should only be used after evaluation by a physician
The regulatory approval of a drug, diagnostic test or medical device should be understood in the context of a whole health system.6 The health system in the USA has a decades-long tradition of governance and oversight of medical decision-making. One of the founding distinctions of regulatory oversight in the USA is the analytic separation between medical research – contributing to generalizable knowledge, and clinical practice – using existing medical knowledge with the intention of improving lives.7 As the FDA approval states, this AD blood test is “intended for patients presenting at a specialized care setting with signs and symptoms of cognitive decline … in diagnosing Alzheimer’s disease”.5 In other words, this blood test has been approved for clinical use. While the primary focus here is on its application in diagnostic practice, its potential role in medical research – particularly in efforts to make academic medicine more sustainable – will also be considered.
How, then, is AD diagnosed? Here, there is another important distinction to draw, this time with regards to the meaning of AD. The Alzheimer’s Association in the USA, the largest patient association with significant global influence in research, policy and advocacy, has a specialist workgroup which defends a “continuum” concept based on the famous amyloid cascade hypothesis of AD.2 According to this definition, AD starts with Aβ accumulation, which triggers downstream biomarkers of tau and neurodegeneration, and ultimately ending in dementia.8 Importantly, these authors believe that AD should be understood in deterministic terms, with the arrival of dementia a matter of time in Aβ-positive individuals, even though the timescale between Aβ accumulation and cognitive decline remains unspecified and would happen after death due to other causes in many or even most people. However, the authors argue that “mortality from unrelated causes should not be a criterion used to define what is and what is not a disease”.8
Global estimates suggest that over 400 million people are on the AD continuum,9 that is, they are amyloid-positive and have AA-2024-defined biological AD (bAD), even though most of them will never develop dementia during their lifetime.10 Hypothetically, these new blood tests could be used to diagnose bAD in more than 400 million people, with the aim of offering a significant proportion of them anti-Aβ antibody therapy to reduce the risk of future cognitive decline. We will argue in the following paragraphs that this would be a mistake because bAD does not meet the criterion of clinical relevance to current and future patients.
The authors of the AA-2024 diagnostic criteria themselves argue that “AD can be diagnosed in asymptomatic individuals, but we do not believe this should be done for clinical purposes at this time” (though the authors also state that this recommendation “would change in the future if disease-targeted therapies, currently being evaluated in trials, demonstrate benefit in preventing cognitive decline and are approved for clinical use in individuals with preclinical AD”).8 Therefore, to maximize the clinical relevance of blood tests in AD, a different definition of AD will have to be used. The International Working Group 2024 Clinical-Biological Construct of Alzheimer Disease (IWG-2024) is grounded in a probabilistic, rather than deterministic, relationship between amyloid accumulation and the clinical state of cognitive decline.11 The IWG-2024 argues that symptoms of a neurocognitive disorder and AD biomarkers are both required to establish a diagnosis of AD.11 The neurocognitive disorder is typically characterized by progressive amnesia, language impairment (aphasia), and behavioral dysregulation, although atypical forms also exist. For most people, accumulation of AD biomarkers alone is a state of risk for AD: Aβ+ individuals are at risk of AD, and according to IWG-2024, the term “preclinical AD” is reserved for people with genetic risk profiles that make dementia “in the near future” the exception rather than the rule for those people.11
Only the IWG definition of AD can ensure the clinical relevance of blood tests in clinical practice, i.e., their use only after expert evaluation.12 A blood test on its own without knowing someone’s cognitive status will not provide clinically meaningful information to those who need it,13 and asymptomatic bAD could lead to psychosocial harms, including legal and financial consequences, and unnecessary treatment.11 Primary care physicians should also be wary of direct-to-consumer promotion of these blood tests which may brush over the issue of clinical relevance in the name of access.12 Commercial entities thus have an obligation to avoid overstating the clinical relevance of blood tests for asymptomatic people.14
Finally, it is vital that the diagnostic performance of blood tests is confirmed in diverse, global populations.15 Inconclusive test results (up to 1 in 5) should motivate more sophisticated imaging technologies to enhance diagnostic accuracy. Now that we have established clinical relevance as a necessary safeguarding criterion for the use of blood tests in confirming diagnosis of AD, we can now explore the 2 potential benefits they offer to global health: improved accessibility to diagnostic confirmation and more sustainable healthcare actions.
Confirmation of AD diagnosis is currently high-tech, high-cost, and low-accessibility: Blood biomarkers could improve access to diagnostic confirmation
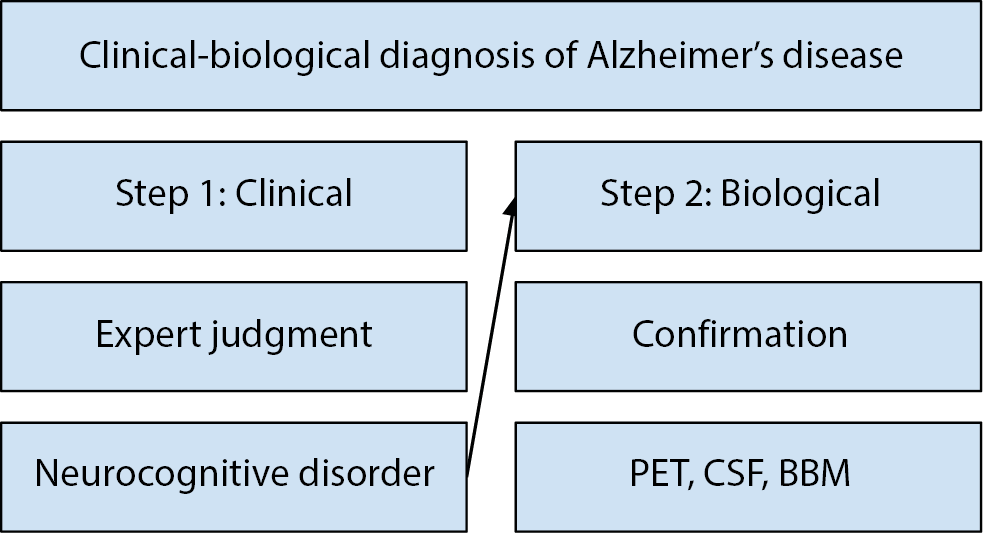
The FDA’s language to describe their approval and the IWG definition of AD lead us to the following claims. Blood tests do not answer the question: Does this asymptomatic person have AD? They instead provide a (high-probability) answer to the question: Is this person’s neurocognitive disorder due to AD? Thus, all that blood biomarkers can currently do is confirm or rule out a diagnosis of AD as part of a (simplified) 2-step clinical-biological process (Figure 1) that is required to establish diagnosis of AD according to best practices.16
Both Step 1 and Step 2 suffer from low access both within countries (e.g., rural vs urban settings), including high-income countries,17 and between countries. The majority of over 55 million cases of dementia worldwide are undiagnosed, though in richer countries, this trend is reducing.18 Thus, investing in human expertise to establish cases of neurocognitive disorder is arguably the most important step that global medicine can take to improve access to diagnosis of AD and dementia worldwide, rather than overly focusing on technological development.19 Blood-based biomarkers of AD cannot directly bolster this process due to the need for robust health systems to perform the expert clinical work of Step 1 that requires years of trying in neuropsychological assessment.15
As for Step 2, prior to the approval of AD-BBMs, AD diagnosis has required detecting the presence of pathophysiologic biomarkers with high specificity for post-mortem neuropathology, including amyloid PET scans and CSF concentrations of amyloid and tau proteins via lumbar puncture or “spinal tap”. Topographic biomarkers are also used to study the regional consequences of AD pathology, such as regional hypometabolism on fluorodeoxyglucose (FDG)-PET, tau PET, and atrophy on structural magnetic resonance imaging (MRI), which can be used to measure disease progression.16
Globally, Step 2 of the AD diagnostic process also suffers from low accessibility, because neither PET scanners nor CSF lumbar punctures are available in many countries. Even in high-income countries where specialist infrastructure exists, the average time from symptom onset to an established diagnosis is approx. 3.5 years.20 Moreover, CSF punctures involve some degree of invasiveness and significant rates of patient refusal in global contexts, and there are important contraindications for use in AD.21
Furthermore, cost is an important factor. In the USA, PET scans cost at least 5,000 USD and are not reimbursed by medical insurance except when performed in research contexts.22 Lumbar punctures cost approx. 1,000 USD and are more likely to be reimbursed; therefore, they are used more frequently in routine clinical practice. But for memory clinics, performing lumbar punctures is expensive due to the staff required for the long procedure.23 While blood tests are thought to be marketed at 500–1,000 USD24 competing tests are deliberately being designed to lower costs.25 In the long term, it is likely that blood tests will remain the most cost-effective option, but more universal, equitable access will only be attained with insurance coverage.26
Thus, diagnostic confirmation of AD is currently high-tech, high-cost, low-accessibility. The overall low accessibility of AD diagnosis worldwide is a barrier to the right to healthcare, the founding mission of the WHO.27 If – and only if – used in conjunction with human expertise to establish clinical-biological AD, access to BBM-ADs worldwide could be a boon for accessibility to timely diagnosis by reducing technological, financial and time barriers to achieve the goal of timely diagnosis of AD.28
In research and practice, blood tests for use in Alzheimer’s disease could be a step towards more sustainable healthcare
Current healthcare practices take place within a period of human-exacerbated climate change. Climate change is arguably the greatest health threat facing humanity across the global population, and health impacts will follow and further exacerbate existing gradients of health inequalities facing people with neurological disorders, which are the most burdensome diseases worldwide, affecting around 3.4 billion people.29, 30 The effects of climate change exacerbate ill health across the spectrum of neurodevelopmental, neurological and neurodegenerative diseases.31, 32 There is an emerging brain-climate research program to study the vulnerability of the human brain to the effects of climate change.33
But health research and clinical practice themselves are a significant source of carbon emissions, due to wet laboratory and clinical research, diagnostics, therapeutics and anesthetics, and computational research.34, 35 It is now recognized that there is a major need for sustainable healthcare practices in neurology.32 However, as mentioned in Section 1, healthcare systems involve both clinical practice (e.g., diagnostics) and research, the latter meaning the creation of generalizable knowledge about health and disease.7 Thus, awareness of climate change should lead us to think of both research and clinical practice in the light of a “healthcare-harm” trade-off: Energy-intensive actions in both practice and research actually contribute to worsened emissions and climate change, and thus worsened (brain) health worldwide. Thus, AD-BBMs could be used to make this healthcare-harm trade-off more favorable by making diagnostic practices and research projects more environmentally sustainable.
Let us take the example of imaging and AD diagnosis and calculate approximate carbon footprints per patient. PET scanners used to confirm AD diagnosis have an annual energy consumption of approx. 10–11 4-person households, leading to the annual emission of approx. 15 tons of carbon dioxide (CO2), and they cannot usefully be switched off due to the hours-long process of recalibration for routine use.36 Excluding all of the costs of making a PET scanner as well as the indirect energy costs during the 2–3 h period of a patient being in the hospital (e.g., heating and lighting) for a PET scan, let us focus just on the amyloid PET scan itself, which takes around 30 min. Assuming scanner availability for a 40-h working week in non-hospitalized (i.e., new) patients, approx. 80 patients could be scanned per week. Assuming some operational time costs due to preparing the scanner for different patients, let us say that a single PET scanner can be used to scan 70 patients per week, for 50 weeks of the year to allow for holidays, meaning 70 × 50 = 3,500 patients per year. Dividing the 15,000 kg of annual emissions by 3,500 patients leaves us with approx. 4.25 kg of CO2 emissions per new patient scanned in a PET scanner. Conversely, a full blood test, leads to the emission of approx. 116 g of CO2, accounting for all consumables, associated waste for blood draw, and electricity and water use for laboratory analyses.37 A rough estimate indicates that a BBM-AD blood test produces approx. ×36 fewer CO2 emissions than a PET scan (4,250/116).
How about the carbon footprint of lumbar punctures? Once more, we will not calculate indirect emissions due to a longer procedure. However, no specific calculations exist for the carbon footprint of lumbar punctures. Related data from anesthesia and surgical interventions provide a rough benchmark, as lumbar punctures require both anesthetic agents and consumables. The anesthetics themselves have a low median converted carbon footprint, e.g., of approx. 75 g,38 whereas the footprint of the surgical consumables (i.e., a spinal anesthesia administration set) and the preparatory consumables (e.g., gowns) are closer to a kilogram.39 Given the 116 g CO2 footprint of a full blood draw, it can be reasonably stated that AD-BBMs generate approx. 10-fold lower CO2 emissions compared with lumbar punctures.
These rough estimates suggest that AD-BBMs have an approx. 10-fold and 36-fold lower carbon footprint compared with CSF lumbar puncture and amyloid PET scanning, respectively. This means that they can increase resource stewardship40 and make for more sustainable healthcare actions when used in line with best practice recommendations.
This could also include healthcare research, for instance, to replace PET in low-precision studies including exploratory longitudinal studies, pilot intervention studies, feasibility studies, and proof-of-concept studies. However, the scope of AD-BBMs is likely to be more limited to research and diagnostic practice, as they cannot achieve other goals of medical imaging, e.g., the use of precision MRI in monitoring the side effects of current and future treatments for AD. It is vital that the sustainability gains from the lower carbon footprint of blood tests do not lead to their misuse in diagnostic practice, which could lead to a loss of those gains in reduced emissions if they are used excessively.37 There are many instances when blood-tests are overused,40 and given the unmet needs of people with AD, there is the risk that these diagnostic aids contribute to overtesting, particularly if the warnings of the defenders of the clinical-biological definition of AD are not heeded.
Conclusions
A recent analysis found significant evidence gaps for AD-BBMs: Most research focuses on their technical capacity; many other papers tend to promote them in commentaries and reviews; a handful of papers have addressed their diagnostic accuracy, whereas hardly any have studied their impact on patients and society.41 These gaps, alongside the revised biological criteria for AD proposed by the Alzheimer’s Association in 2024, suggest the possibility of widespread, clinically irrelevant use of AD-BBMs. This bias should be corrected by re-focusing research on findings that meet the criterion of relevance to current and future patients.
Nevertheless, if their relevance to global populations is confirmed, and if they are used wisely in conjunction with clinical expertise, AD-BBMs could be an accessible, sustainable front-line tool to increase the coverage of confirmation of timely AD diagnosis. Their use will require a logic of trade-offs to maximize gains and limit harms for current and future patients.
Use of AI and AI-assisted technologies
Not applicable.