Abstract
Systemic lupus erythematosus (SLE) is a chronic, autoimmune inflammatory disease with a multisystem manifestation and a variety of clinical symptoms. Over the last decades, the prognosis and life expectancy of patients with SLE improved significantly due to the implementation of corticosteroids combined with immunosuppressive agents. Nevertheless, the use of these medications is often associated with the occurrence of serious side effects and additional deterioration of organ function. Therefore, developing and implementing novel therapies that are both safer and more effective in managing disease is crucial. For a long time, European Alliance of Associations for Rheumatology (EULAR) recommended only 2 biological agents in the treatment of SLE: belimumab and rituximab. However, in 2023, anifrolumab, an interferon (IFN) receptor inhibitor, and voclosporin, a novel calcineurin inhibitor, appeared in new SLE treatment guidelines. In addition, several biological agents are targeting different cells or cytokines that are being evaluated in phase II and III clinical trials. Apart from that, experimental therapies such as targeting of plasma cells, chimeric antigen receptor T-cell therapy (CAR-T) or stem cell transplantation appear promising in the treatment of the severe forms of SLE.
Key words: systemic lupus erythematosus, calcineurin inhibitors, biological therapy, monoclonal antibodies
Introduction
Systemic lupus erythematosus (SLE) is a chronic, multifaceted autoimmune disease with a broad range of symptoms and a variable prognosis. Early diagnosis and initiation of the treatment are crucial to alleviate symptoms and reduce mortality.1 The development of new medications is necessary to further extend the patients lifespan and improve their quality of life. Systemic lupus erythematosus affects primarily women of reproductive age, with female to male ratio of 9:1.2, 3 Its incidence for the global population is estimated to be 5.14 per 100,000 person/years.4
Common SLE manifestations often include fatigue, weight loss, arthritis, and skin rashes, especially butterfly-shaped rash, which spreads across cheeks and nose (malar rash).5 Affected individuals commonly experience photosensitivity, provoking skin reactions upon sun exposure. Throughout the disease, lesions in internal organs can be seen. Lupus nephritis (LN) is one of the most frequent severe organ manifestations of SLE, carrying a substantial morbidity and mortality risk, with approx. 20% of patients advancing to end-stage renal disease (ESRD).6, 7 Although less common, neuropsychiatric SLE manifests across a spectrum of presentations, ranging from mild cognitive dysfunction to severe psychosis.8 Furthermore, SLE may give a rise to manifestations in pulmonary, gastrointestinal and cardiovascular system, while also affecting hematopoiesis.
Although the short-term and median outcomes of SLE patients have improved over the past decade, the long-term prognosis is still unfavorable.9, 10 The course of the disease is burdened by comorbidities in addition to the side effects of the treatment used.11, 12 These include cardiovascular disease such as arteriosclerosis, hypertension, osteoporosis, and increased frequency of infections.13 The prevalence of psychiatric disorders such as depression and anxiety is significantly higher compared to the general population.14 Frequently occurring fatigue interferes with the ability to perform routine daily activities.15 A substantial number of individuals are forced to reduce their work hours or even retire.16
Systemic lupus erythematosus is associated with the production of autoantibodies targeted mainly against autoantigens coming from cell nuclei and the formation of immune complexes that cause damage to various organs. However, the exact cause of the disease is still unknown. It is being suggested that genetic, environmental and hormonal factors play a role in self-immunization.17, 18 Its multifactorial pathogenesis together with its variable clinical phenotypes pose a challenge to the treatment. However, the rising knowledge of the pathological pathways taking part in the disease progression allows us to introduce new therapeutic solutions.
Objectives
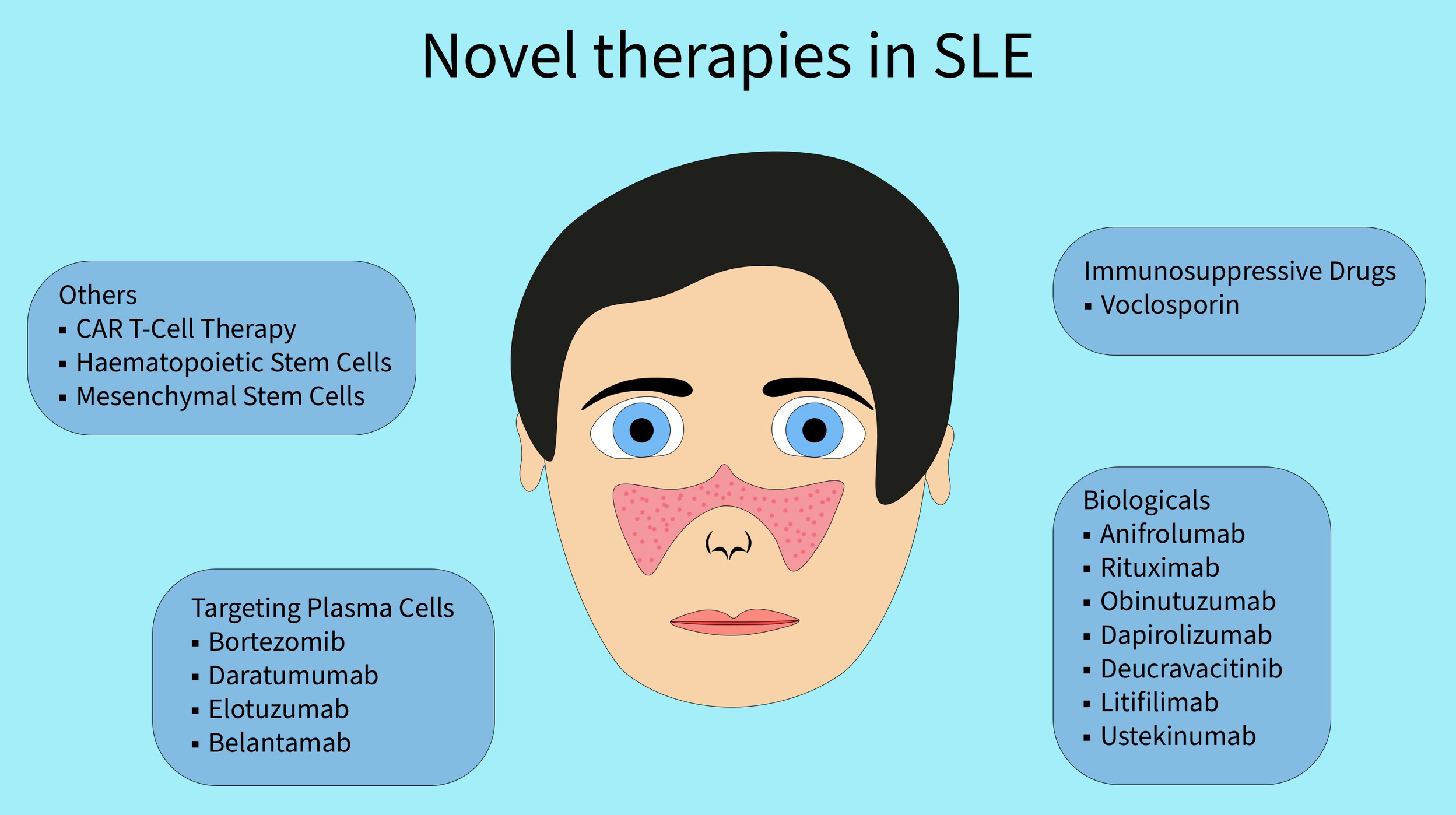
Due to the increasing incidence of SLE and the growing number of patients who do not respond to previously used medications, this systematic review aims to present newly introduced SLE therapies as well as treatments with potential for broader use in the future (Figure 1).
Materials and methods
The literature search used PubMed, Embase and Google Scholar databases, as well as references from relevant articles and internet sources. Search terms included “SLE novel therapies”, “SLE biological medications”, “SLE future therapies”, “SLE voclosporin”, “SLE anifrolumab”, “SLE obinutuzumab”, “SLE dapirolizumab”, “SLE deucravacitinib”, “SLE ustekinumab”, “SLE litifilimab”, “SLE plasma cells targeting”, “SLE CAR-T”, and “SLE stem cell transplant”. The authors screened the titles and abstracts to identify relevant articles, with the last literature search performed on May 5, 2024. Finally, we have included 116 studies eligible for our review.
Immunosuppressive medications
Voclosporin
Calcineurin inhibitors (CNIs) are immunosuppressive medications widely used in transplantology and the treatment of autoimmune diseases. Calcineurin inhibitors disrupt the intrinsic calcium signaling pathway, ultimately leading to decreased T-cells activation, proliferation and differentiation. What is more, CNIs act in nephron podocytes to stabilize the actin cytoskeleton, thereby exerting an antiproteinuric effect. This unique characteristic of CNI makes them ideal candidates for the treatment of autoimmune glomerulonephritis, including LN.
Voclosporin is a novel CNI indicated for the treatment of adult patients with active class III, IV and V LN, and in combination with mycophenolate mofetil (MMF). Compared to cyclosporin, low-dose voclosporin seems to have a lower nephrotoxicity, and compared to tacrolimus, a lower diabetogenic effect.19 AURORA 1 study assessed the efficacy and safety of voclosporin compared to placebo in patients with biopsy-confirmed LN over the course of 2 years. Voclosporin in combination with MMF and low-dose steroids caused clinically significant complete renal responses compared to placebo (41% vs 23% of patients), with a comparable rate of serious adverse events (21% in both groups).20 The continuation of this study, AURORA 2, further evaluated the long-term efficacy with adverse effects. Furthermore, it determined biochemical and hematological outcomes. Over a 3-year follow-up, the rate of adverse events was similar to that seen in the AURORA 1 study. Hypertension and decreased glomerular filtration rate (GFR) were observed more frequently with voclosporin. However, the mean corrected estimated GFR (eGFR) was within the normal range and stable in both groups. A complete renal response occurred in 50.9% of patients in the treatment group compared to 39.0% in the placebo group.21 It has been reported that the combination of voclosporin and MMF does not require a change in the dosage of MMF.22
Targeted therapies
Anifrolumab
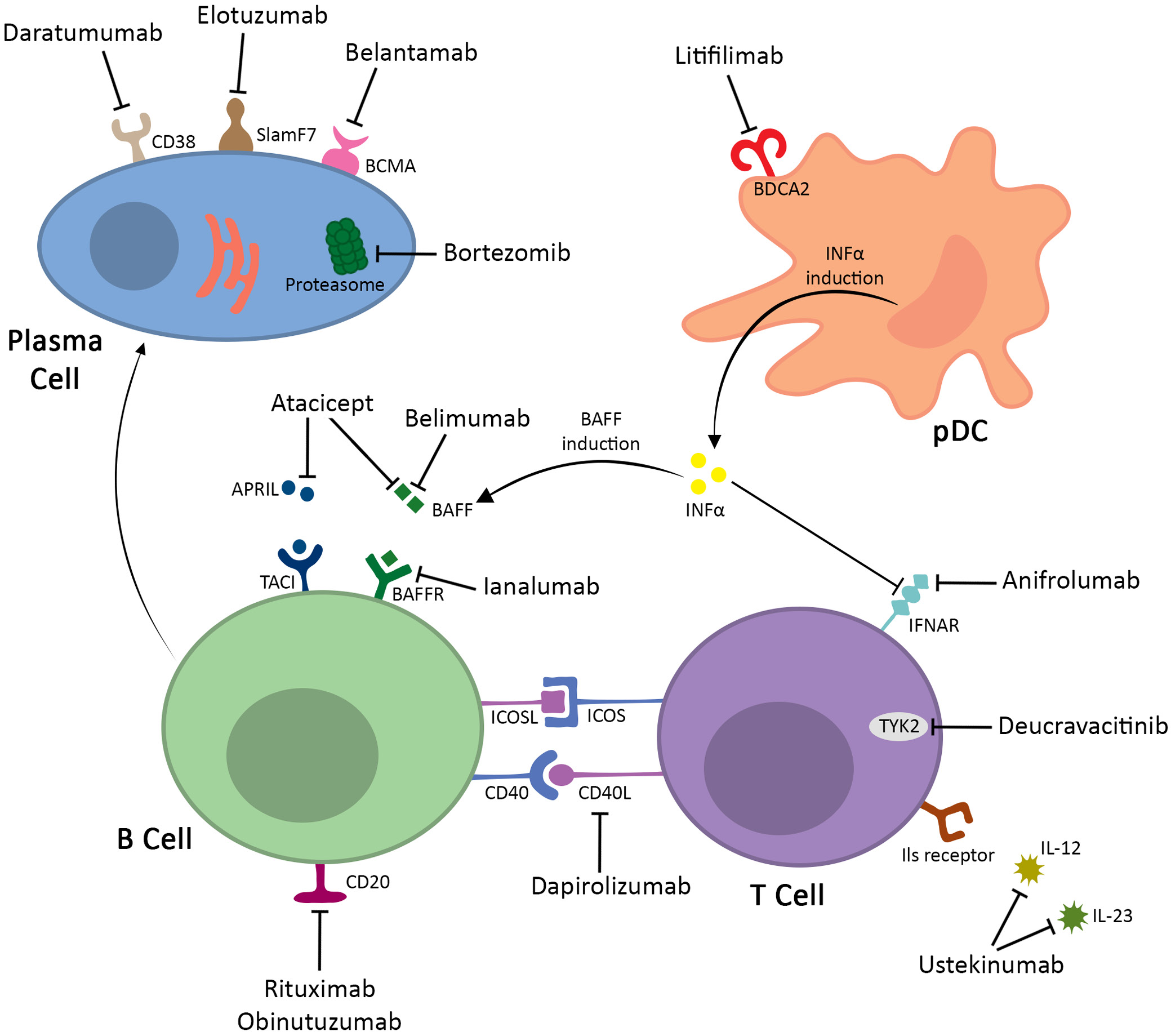
The interferon (IFN) pathway has attracted a lot of attention as one of the key factors in pathogenesis of SLE and as a promising aim of novel treatment methods.23 The “type I IFN signature”, or overexpression of gene transcripts in the IFN pathway, is a hallmark of SLE patients.24 This, in turn, results in the dysfunction of peripheral tolerance mechanisms. The interferon promotes activation of Th cells, improves plasmacytoid dendritic cells abilities to present antigens, and induces the production of various cytokines.17 The IFN overproduction is probably mediated by exposure of plasmacytoid dendritic cells (pDC) to serum immune complexes and increased neutrophil extracellular trap (NET) formation, while simultaneously reducing the ability to degrade them. Other proposed triggers are bacterial and viral infections, dysregulation in gut microbiome and increased estrogen concentrations. Over half the loci associated with SLE encode proteins related to cell IFN production or response.25
Anifrolumab is becoming a promising therapeutic option for patients with SLE and is currently used in the therapy of moderate-to-severe SLE.26 Anifrolumab is a human monoclonal immunoglobulin 1 (IgG1) antibody to the type I IFN receptor subunit 1 (IFNAR1) that binds to type I interferon receptors blocking their activation and induction of interferon-related genes. Numerous studies have proven that type I IFN system plays a crucial role in the SLE etiopathogenesis.27, 28 Sandling et al. identified an increased expression of type I IFN-inducible genes in SLE patients, referred to as the ‘IFN signature.’ Additionally, Beachler et al. demonstrated that polymorphisms in genes such as IKBKE and IL8 are associated with dysregulated type I IFN signaling, potentially increasing susceptibility to SLE by altering inflammatory pathways.29, 30 In addition, a causative role of the type I IFN system in the development of SLE has been demonstrated, as individuals treated with IFN-α developed this disease that was indistinguishable from the naturally occurring disease.31 Type I IFNs are a family of cytokines that include 13 subtypes of IFN-α, as well as IFNβ, IFNε, IFNκ, and IFNω. All of these type I IFNs initiate a signaling cascade by binding to the receptor complex composed of IFNAR1 and IFNAR2 and thus inducing conformational changes in these receptors.32, 33, 34 This leads to activation by the tyrosine kinases JAK1, which interacts with IFNAR2, and TYK2, which in turn interacts with IFNAR1. Janus kinases activate STAT1 and STAT2, which then initiate gene transcription, specifically IFN-stimulated response elements found in IFN-stimulated genes. Hence, any alternations in type I IFN signaling pathway might disturb homeostasis and prolong the biological effects of IFNs, which consequently might cause uncontrolled destructive effects observed in SLE.
Following the newest European Alliance of Associations for Rheumatology (EULAR) 2023 recommendations, the first-line treatment of SLE is hydroxychloroquine at a target dose of 5 mg/kg real body weight/day. Glucocorticoids are recommended a maintenance dose ≤5 mg/day (prednisone equivalent), and if possible, they should be withdrawn. In patients with moderate-to-severe disease, there might be considered pulses of intravenous methylprednisolone. In patients not responding to hydroxychloroquine alone or in combination with glucocorticoids or patients unable to reduce their dose below the levels acceptable for chronic use, the use of immunomodulating agents or mycophenolate and/or biological agents, such as belimumab or anifrolumab, should be considered.35
Biological agents are critical for some patients to better control their disease, and that is why it is so important to develop and research these drugs. Belimumab and anifrolumab have demonstrated efficacy in controlling disease activity and allowing GC dose reductions. In 2021, the U.S. Food and Drug Administration (FDA) approved the use of anifrolumab in the SLE treatment. In contrast, belimumab has been used in clinical practice for over a decade.32, 33 There is no given hierarchy between anifrolumab and belimumab in the EULAR 2023 recommendations as these 2 drugs have not been compared in the head-to-head trials. Apart from reducing the disease activity, anifrolumab has the potential to reduce the dose of corticosteroids used to treat SLE. In TULIP 1 and TULIP 2 trials, sustained corticosteroids tapering was achieved in 52% of patients in the anifrolumab group and in 32% of patients in the placebo group. In addition, it was possible to reduce the cumulative corticosteroid dose by 32% in taper responders in the anifrolumab group, the blood pressure was reduced and anifrolumab group experienced fewer side effects.36 What is more, in patients with moderate-to-severe SLE, anifrolumab has been demonstrated to reduce the incidence of flares. In patients who achieved sustained corticosteroids reduction, 40.0% did not experience flares while on anifrolumab compared to 17.3% receiving placebo.37 In the long-term extension (LTE) of the TULIP 1 and TULIP 2 trials, the risk of non-opportunistic infections was similar between the anifrolumab and placebo groups over a 3-year observation period, and the risk of serious adverse events was lower in the treated group.38 The potential of anifrolumab in patients with severe LN was assessed in phase II randomized trial. In the intensified regimen group (3 initial doses of 900 mg followed by 300 mg maintenance doses), 45.5% of patients achieved a complete renal response (CRR), compared to 31.1% in the placebo group. Furthermore, sustained corticosteroid reduction was observed in 55.6% of patients in the anifrolumab group compared to 33.3% in the placebo group.39 Long-term extension provided similar results, also showing the safety of the treatment, with adverse event rates of 6.9% and 8.7% in the anifrolumab and placebo groups, respectively.40
Obinutuzumab
The cell-surface antigen CD20, expressed on mature B cells and most malignant B cells, is an excellent target for the treatment of B-cell malignancies as well as autoimmune disorders.41 Numerous studies have shown that B-cell depletion therapy with anti-CD20 monoclonal antibodies (mAbs), such as rituximab, have notably improved symptoms and clinical remission in patients with rheumatoid arthritis.42 Moreover, administration of anti-CD20 mAb in the population of SLE patients has been associated with a substantial decrease in plasma cell population43 subsequently reducing several SLE antibodies including anti-dsDNA, anti-nucleosome and anti-cardiolipin.44 Anti-CD20 antibodies can exhibit functional activity in 3 different ways: signaling in target cells leading to growth inhibition and non-classical apoptosis, described as “direct cell death”, complement-dependent cytotoxicity (CDC), and antibody-dependent cellular cytotoxicity (ADCC), mediated by cells displaying Fcγ receptors (FcγRs).41, 45
Two types of effector function profiles of CD20 antibodies have been described, referred to as type I and type II. They have been distinguished by the CD20 epitope, to which the antibodies bind and/or their binding mode.46 According to the study by Cragg and Glennie, the therapeutic efficacy of type I mAbs is directly related to the classical pathway of complement activation through their binding to the C1q component. On the other hand, type II mAbs do not utilize complement or NK cells for their function. Instead, their therapeutic activity is achieved through potent induction of direct apoptosis.46
In randomized controlled trials, rituximab, a type I mAb, was not effective in the treatment of SLE and LN.47 However, observational studies, large retrospective studies and meta-analyses of observational studies have shown efficacy in the treatment of SLE and LN with complete response estimates of 46–57% and 36–51%, respectively.48 Furthermore, a study conducted by van der Kolk et al. has shown that most of the side effects of anti-CD20 type I mAbs treatment were correlated with the activation of complement.49 Therefore, it is suggested that type II antibodies could potentially offer the benefit of lower toxicity. These beneficial qualities have led to increased interest in anti-CD20 type II mAbs, resulting in the development of new monoclonal antibodies, such as obinutuzumab.
Obinutuzumab, also known as GA101, is a humanized, Fc-engineered type II IgG1 antibody targeted against CD20. It was originally engineered and characterized by Mössner et al. in 2010.50 Obinutuzumab was compared in the preclinical studies with another anti-CD20 mAb, rituximab, a humanized, chimeric type I IgG1 antibody.51 GA101 showed in vivo efficacy superior to rituximab in all tested parameters. In the aggressive human B-cell lymphoma xenograft models, tumor growth inhibition was more effectively achieved by obinutuzumab compared to rituximab. Moreover, GA101 exhibited a dose-dependent enhancement in performance leading to a complete tumor regression at a dose of 30 mg/kg, while rituximab failed to achieve this at any dose used. In addition, obinutuzumab demonstrated superior B-cell depleting activity in the blood in the cynomolgus monkeys in comparison with rituximab. Studies also showed that B-cell depletion by GA101 was greater in spleen and lymph nodes.50 According to the study by Beers et al., type II anti-CD20 antibody complexes tend to remain on the B-cell surface for extended periods of time, resulting in a more efficient depletion of the B-cells compared with type I. This could possibly be linked to a more potent effectiveness of obinutuzumab, as indicated in the previous study.52 Those promising results from the preclinical studies suggest that obinutuzumab has the potential to be used in the SLE treatment.
A phase II trial (NOBILITY), conducted in 2021, compared obinutuzumab with a placebo in the management of LN in association with standard therapies involving mycophenolate mofetil and steroids. A total of 125 patients with SLE, aged between 18 and 75 years and presenting with class III or IV LN, were included in the study. The participants were randomly divided into 2 equal groups to receive either obinutuzumab 1,000 mg or placebo infusions on day 1 and week 2, 24 and 26. A total of 115 patients completed 52 weeks and 103 patients completed 104 weeks of follow-up. At week 52 of the protocol, the primary endpoint, CRR, was achieved to a greater extent by the obinutuzumab group (35%) compared to the placebo group (23%). Although the last dose of the drug was administered at week 26, the advantage persisted throughout the study reaching 41% of CRR in the obinutuzumab group and remaining unchanged in the placebo group. Moreover, the results in the test group outperformed the results in the control group, reaching a higher overall renal response (ORR), consisting of CRR and partial renal response (PRR), with a rate of 55% compared to 35% in the control group at week 56, and 54% compared to 29% at week 104. The positive effects of obinutuzumab were especially noticeable in patients with class IV LN and those with a baseline urine protein-to-creatinine ratio (UPCR) ≥3. In addition, obinutuzumab led to a more substantial increase in C3 and C4 levels as well as improvement in eGFR. Moreover, anti-dsDNA antibody levels decreased significantly with applied treatment. Compared to placebo, UPCR levels also showed a greater reduction. In terms of safety, the use of obinutuzmab did not show any correlation with an increase in serious adverse events over a period of 2 years, nor with serious infections or fatalities. Furthermore, there were no instances of severe reactions related to the infusion or severe cases of thrombocytopenia or neutropenia.53 The data gathered has prompted the initiation of a phase III trial (REGENCY) to test obinutuzumab in patients with class III or IV LN.
Another study, conducted by Arnold et al., verified the validity of obinutuzumab in SLE patients with secondary non-depletion nonresponse (2NDNR) to rituximab. Nine patients who had been previously treated with cycles of rituximab 2 × 1,000 mg and developed 2NDNR were switched to obinutuzumab 2 × 1,000 mg infusions alongside methylprednisolone 100 mg. Six months after the treatment, there were substantial reductions in median Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) and total British Isles Lupus Assessment Group 2004 (BILAG-2004) score as well as significant improvements in C3 and dsDNA levels. Six patients achieved complete B-cell depletion, of whom 4 reached a SLE low disease activity state (LLDAS) with reduced methylprednisolone dosage. Also, no adverse, infusion-related events were observed.54
In addition, the efficacy of obinutuzumab in renal and non-renal SLE is currently being evaluated in a phase III trial (OBILUP, NCT04702256) and a phase III trial (ALLEGORY, NCT04963296), both launched in 2021.
Dapirolizumab
CD40 ligand (CD40L), mainly found on activated T lymphocytes and platelets, together with its receptor CD40, are involved in regulating interactions between T cells and other cells. These actions lead to increased B-cell proliferation and differentiation, antibody production, as well as the formation of germinal centers in lymph nodes. Given its pivotal role in activating the immune system, and thus influencing the development of SLE, the CD40 ligand has become a potential target for the treatment of this condition.54, 55
Dapirolizumab is a polyethylene glycol-conjugated antigen-binding (Fab′) fragment that targets CD40L.56 Unlike its predecessors directed against CD40L, dapirolizumab lacks a functional Fc domain, which has been reported to carry a risk of thromboembolism.56 In order to verify the safety of dapirolizumab, 2 phase I clinical trials were conducted. In the 1st one, healthy volunteers and SLE patients were administered a single dose of dapirolizumab or placebo. The 2nd trial examined the response to receiving multiple doses of the drug. In the final analysis, in both cases, dapirolizumab was shown to be well-tolerated and no thromboembolic events occurred.57, 58
A randomized, placebo-controlled phase II clinical trial of dapirolizumab in patients with active SLE has failed to demonstrate a pre-specified dose-response relationship. One hundred and eighty-two patients were randomly assigned to receive placebo or dapirolizumab at a dose of 6 mg, 24 mg or 45 mg every 4 weeks until week 20. The overall treatment efficacy was assessed with British Isles Lupus Assessment Group–based Composite Lupus Assessment (BICLA) at week 24. Despite not reaching the primary endpoint, dapirolizumab-treated patients experienced significant improvements in Systemic Lupus Erythematosus Responder Index (SRI-4), BICLA, SLEDAI-2K, Physician Global Assessment (PGA), BILAG, and Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI) scores relative to the placebo group. Moreover, the tested drug appeared to effectively reduce anti-dsDNA levels and increase C3 and C4 levels. In addition, dapirolizumab lowered the risk of severe flares compared to placebo (5 vs 7). Considering the safety of dapirolizumab, it was rated as acceptable, with a similar incidence of adverse events among all groups. Similarly to phase I trials, treatment with dapirolizumab did not increase the risk of thromboembolism. On the other hand, the tested drug raised the frequency of infections, mainly those affecting the upper respiratory tract.59 The overall results of this study have contributed to a phase III clinical trial to further evaluate the efficacy of dapirolizumab.
Deucravacitinib
Janus kinases (JAKs) are a group of enzymes comprised of JAK1, JAK2, JAK3, and TYK2, involved in transmitting information from the cytokine receptors to the cells. The JAKs phosphorylate STATs (signal transducers and activators of transcription), enabling them to form dimers and translocate into the cell nucleus, where they bind to DNA and activate the transcription of specific genes. This JAK-STAT pathway plays a crucial role in hematopoiesis, inflammation and immune response.60, 61 TYK2 protein binds with JAK1 and JAK2 to mediate the signaling of several cytokines involved in the pathogenesis of SLE, especially type I IFNs, interleukin (IL)-10, IL-12 and IL-23.62, 63, 64
Deucravacitinib is an oral, allosteric, highly selective inhibitor of TYK2. Unlike the other kinase inhibitors, it binds to the catalytically inactive regulatory pseudokinase JH2 domain of the TYK2, thereby blocking the enzyme in its inactive state and preventing downstream signal transduction.65, 66, 67 The FDA has approved deucravacitinib in the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. The drug’s safety profile was documented by Catlett et al. in a phase I clinical trial involving 100 healthy participants, 75 of whom received the medication. Deucravacitinib was rapidly absorbed and had a half-time of 8–15 h. The drug was found to be safe and well tolerated. No serious adverse events (AEs) were reported. The incidence of non-serious AEs in test group (64%) was not higher than in the placebo group (68%). The most reported adverse events were headache, nausea, rash, acne, and upper respiratory tract infections.68
A randomized, double-blind, placebo-controlled phase II clinical trial (PAISLEY) tested the efficacy and safety of deucravacitinib in adult patients with active SLE with SLEDAI-2K score ≥6 and at least 1 BILAG A or >2 BILAG B manifestations from the musculoskeletal or mucocutaneous domain. Respondents were randomly divided into 4 equal groups receiving deucravacitinib 3 mg twice daily, 6 mg twice daily, 12 mg once daily, or placebo for 48 weeks. The primary endpoint of SRI-4 was evaluated at week 32. A significantly higher response rate of SRI-4 was observed in the group taking 3 mg deucravacitinib than placebo (58.2% vs 34.4%). In the 6 mg and 12 mg dose groups, the primary endpoint was achieved by 49.5% and 44.9% of patients, respectively. Moreover, this correlation was maintained among all groups until the end of the study at week 48. Secondary endpoints assessed at week 48 were met by more patients in all test groups compared to placebo but only the group taking the 3 mg dose achieved a statistically significant difference. Deucravacitinib demonstrated higher BICLA responses (47.3% vs 25.6%), the organ-specific end points for skin (CLASI-50 response, 69.6% vs 16.7%), the treat-to-target end point LLDAS (36.3% vs 13.3%), and a substantial mean change from baseline in the joint count (–8.9 vs –7.6). Furthermore, there was a noticeable enhancement in the C3 and C4 levels, along with a reduction in anti-dsDNA antibodies in patients who were administered deucravacitinib throughout the study. Additionally, a significant decrease in the expression of the IFN gene was observed starting from the 4th week. As for safety, adverse events occurred at similar levels in the test groups as in the placebo group, with the most frequent being upper respiratory tract infections, headaches, nasopharyngitis, and urinary tract infections. Nonetheless, acne and rash occurred more frequently with deucravacitinib treatment, with a significantly higher percentage at 6 mg and 12 mg dose. No deaths, systemic opportunistic infections, active tuberculosis, hematologic malignancies, or major cardiovascular incidents occurred.69 The encouraging outcomes of the PAISLEY trial have prompted the creation of 2 phase III trials (POETYK SLE-1, NCT05617677, and POETYK SLE-2, NCT05620407) that will explore the potential of deucravacitinib in treating extra-renal SLE.
Litifilimab
Plasmacytoid dendritic cells, derived from bone marrow, constitute a specialized subset of DCs.70 They represent a minor part of peripheral blood leukocytes and organized lymphoid tissue that secrete large amounts of type I IFNs in response not only to various bacterial and viral stimuli but also to SLE immune complexes.71, 72 Numerous studies have indicated that patients with SLE have increased levels of pDC in both skin lesions and affected organs, such as the kidneys, putting them in the spotlight for developing new therapies for SLE.73, 74, 75
Litifilimab is a humanized IgG1 mAb that binds to the blood dendritic cell antigen 2 (BDCA2), a receptor expressed on pDC cells.76 As a consequence, litifilimab contributes to a significant suppression of IFN, other cytokines and chemokine production.77 The first phase I clinical trial conducted to evaluate the safety, tolerability and pharmacokinetics of litifilimab involved 54 healthy volunteers and 12 patients with SLE. It consisted of 3 parts, in which volunteers were administered either single or multiple doses of the drug or placebo. Litifilimab was found to be safe and well tolerated, as well as effective in reducing BDCA2 levels on pDCs, lowering CLASI-A scores, inhibiting INF-1 production and normalizing IFN-response markers, including the expression of myxovirus resistance protein A (MxA) in skin lesions.76
Furie et al. continued to further assess the efficacy of litifilimab in patients with SLE and CLE in a 2-part, randomized, placebo-controlled phase II trial. A total of 110 participants were enrolled in part A, which was focused on managing active SLE. Patients were randomized in a 1:1 ratio to receive either a placebo or 450 mg of litifilimab in addition to their standard of care. The primary endpoint based on the reduction from baseline in the number of active joints at week 24 demonstrated superiority of litifilimab over placebo (–15 ±1.2 vs –11.6 ±1.3). In addition, more patients treated with litifilimab achieved a decrease of at least 7 points on the CLASI-A score (56% vs 34%), as well as a greater change in the SLEDAI-2K score. Moreover, the litifilimab group included a higher number of SRI-4 responders in comparison with placebo group (56% vs 29%). However, litifilimab did not appear to significantly reduce the number of SLE-associated autoantibodies or increase C3 and C4 levels.78 Part B focused on the efficacy of litifilimab in the treatment of CLE and included 132 participants. Patients were randomized to receive placebo or litifilimab 50 mg, 150 mg or 450 mg until week 12. Litifilimab treatment demonstrated a significant advantage over placebo in CLASI-A scores resulting in least-squares mean differences of –24.3 percentage points for the 50 mg dose, –33.4 percentage points for the 150 mg dose and –28.0 percentage points for the 450 mg dose compared to placebo at week 16. In terms of safety, litifilimab appeared to be safe and well-tolerated, with more patients experiencing adverse events in the placebo group (68%) compared to the litifilimab group (59%). The most frequent side effects of the tested drug included diarrhea, nasopharyngitis and urinary tract infections.79
Three clinical trials are currently underway: phase III trial (TOPAZ-1), phase III trial (TOPAZ-2) and a 2-part phase II/III trial (AMETHYST) that will provide more information on the efficacy and safety of litifilimab in SLE patients.
Ustekinumab
Interleukin 12 and IL-23 have been identified as crucial cytokines involved in SLE pathogenesis. The former promotes inflammation and triggers the differentiation of Th cells into Th1 cells and stimulates B cells to produce autoantibodies. In addition, IL-12 has a significant function in microbial response by activating NK cells. It consists of a heterodimeric structure composed of 2 subunits, p40 and p35, and is released by monocytes, macrophages and dendritic cells (DCs). Interleukin 23 plays a key role in chronic inflammation. It suppresses the production of IL-2 and is essential for the differentiation of Th cells into Th17 that secrete IL-17 and subsequently induce inflammation by targeting endothelial cells, macrophages, fibroblasts, and keratinocytes. Interleukin 23 has a similar structure to IL-12, composed of a p19 subunit and a shared p40 subunit. It is secreted by antigen-presenting cells, mainly macrophages, DCs and keratinocytes.80 Both IL-23 and IL-12 levels have been found to be notably increased in patients with SLE compared to control groups.81, 82 The associated pathways of the IL-12 and IL-23/Th17 axis in the pathogenesis of SLE have contributed to the development of a new drug targeting these cytokines.83
Ustekinumab is a human IgG1κ monoclonal antibody directed against the p40 shared subunit of IL-12 and IL-23.84 It has been previously approved in the treatment of plaque psoriasis, psoriatic arthritis, Crohn’s disease, and ulcerative colitis.
In a phase II trial conducted by van Vollenhoven et al., 102 participants with seropositive active SLE were randomized (3:2) to either receive 90 mg of ustekinumab or placebo every 8 weeks. The placebo group started receiving ustekinumab 90 mg at week 24. The last dose of the drug was administered to both groups at week 40. The primary endpoint of SRI-4 was evaluated at week 24 and was achieved by 62% patients from the ustekinumab group compared to 33% from the placebo group. In terms of safety, ustekinumab did not increase the risk of adverse events, with the most common being upper respiratory and urinary infections and nasopharyngitis.85
The study was extended to week 120 and 46 participants were enrolled, 29 in the ustekinumab group and 17 in the placebo group, with a final dose at week 104. Interestingly, the SRI-4 response assessed at week 112 was achieved to a greater extent by the placebo crossover group (92%) compared to ustekinumab group (79%). Furthermore, both the ustekinumab and the placebo crossover group had significant improvements in SLEDAI-2K score (92% in both), PGA score (79% and 93%, respectively) and active joint count (86% and 91%, respectively). No deaths, malignancies, opportunistic infections, or tuberculosis cases occurred in the study.84 On the contrary, a phase III trial failed to meet the expectations of the previous study and was terminated as both the primary endpoint; therefore, secondary endpoints were not met.86
Moreover, it appears that administration of ustekinumab may increase the risk of new-onset SLE or its flares. A case report indicated that a 68-year-old patient with chronic plaque psoriasis was started on ustekinumab, resulting in the development of subacute cutaneous lupus erythematosus (SCLE).87
Targeting of plasma cells
Plasma cells are differentiated B-lymphocyte white blood cells capable of secreting immunoglobulin or antibodies. They are divided into long- and short-lived cells.88 B-cell-focused treatments may impact the plasma cell section, specifically plasmablasts, by eliminating plasma cell precursors (rituximab – anti-CD20), inhibiting plasma cell differentiation (belimumab – anti-BAFF, atacicept – anti-BAFF/APRIL) or a combination of both (ianalumab – anti-BAFF receptor).89 However, these therapies generally have no effect on the long-lived plasma cells, as demonstrated on the example of rituximab.90, 91 This shows that plasma cell-directed therapy may become an alternative strategy in the future, particularly for patients who are refractory to the B-lymphocyte-directed therapy. The following strategies are currently under consideration: proteasome inhibition, therapeutic antibodies, chimeric antigen receptor T-cell therapy (CAR-T), or antigen-specific targeting.
Proteasomes function as integral components within a pivotal cellular mechanism, facilitating the regulation of specific protein concentrations and the degradation of misfolded proteins. The identification and targeting of proteins for degradation involves the attachment of a protein known as ubiquitin.92 Bortezomib is the 1st-generation, reversible proteasome inhibitor. It selectively blocks the function of the 26S proteasome, resulting in a lack of proteolysis of the ubiquitin-proteasome complex. This leads to the accumulation of both misfolded and unfolded proteins, which results in the endoplasmic reticulum stress and the unfolded protein response, leading to an increased susceptibility to apoptosis. Proteasome inhibitors also inhibit nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling, an important pathway for long-term plasma cell survival.93
Bortezomib was evaluated in animal models of SLE and has shown efficacy in depleting both short- and long-lived plasma cells in SLE-prone mice. As a consequence, the depletion of the plasma cells producing anti-dsDNA antibodies was observed, as well as the alleviation of nephritis and a significant increase in survival.94 Bortezomib has also been tested in patients with SLE. The study showed a significant reduction in the disease activity, attributable to the therapeutic intervention, characterized by a marked reduction in anti-dsDNA antibodies (approx. 60%), which exceeded the reduction in vaccine-induced protective antibody titers (approx. 30%). There was also a reduction in the population of plasma cells in the peripheral blood and bone marrow (approx. 50%).95, 96 Bortezomib is not specific for plasma cells and therefore causes a number of side effects in the treatment, leading in many cases to the discontinuation of the treatment. Potential side effects include an increased risk of peripheral neuropathy as well as cardiovascular and muscular complications.97 In another, small, randomized trial involving people with SLE, a high rate of the treatment discontinuation due to serious side effects was found. Additionally, contrary to the previously cited study, there was only a minimal effect on dsDNA titers. The change in anti-dsDNA antibody titer did not support the effectiveness of bortezomib as a therapeutic intervention for SLE. Despite this result, the elevated SRI-4 among the treatment group suggests that bortezomib may have the potential to engage mechanisms beyond the suppression of the anti-dsDNA antibody production.98
An alternative approach involves the use of antibodies directed against surface markers that are upregulated at different stages of plasma cell development, a therapeutic strategy currently used in the treatment of multiple myeloma. Notable antibodies in this category include daratumumab (anti-CD38), elotuzumab (anti-SlamF7) and belantamab (anti-B-cell maturation antigen (BCMA)). CD38 and SlamF7 have non-exclusive expression patterns in plasma cells.89 The therapeutic use of SlamF7 expression seems promising in the treatment of SLE. However, targeting CD38- or SlamF7-positive cells should be approached with caution, as this may inadvertently affect other immune cell populations, including B and T regulatory cells in the case of anti-CD38, and NK cells in the case of anti-SlamF7.99, 100 In contrast, BCMA expression is more specific to plasma cells, which has led to various methods for targeting it in the treatment of multiple myeloma. However, the effectiveness of these methods on plasma cells from different sources is still under investigation in this therapeutic approach. Instances of successful therapeutic outcomes in patients with life-threatening, refractory SLE after receiving daratumumab have been documented.101 Nevertheless, it is important to note that their administration resulted in a concomitant reduction in tetanus-specific and total IgG antibodies.
Chimeric antigen receptor T-cell therapy
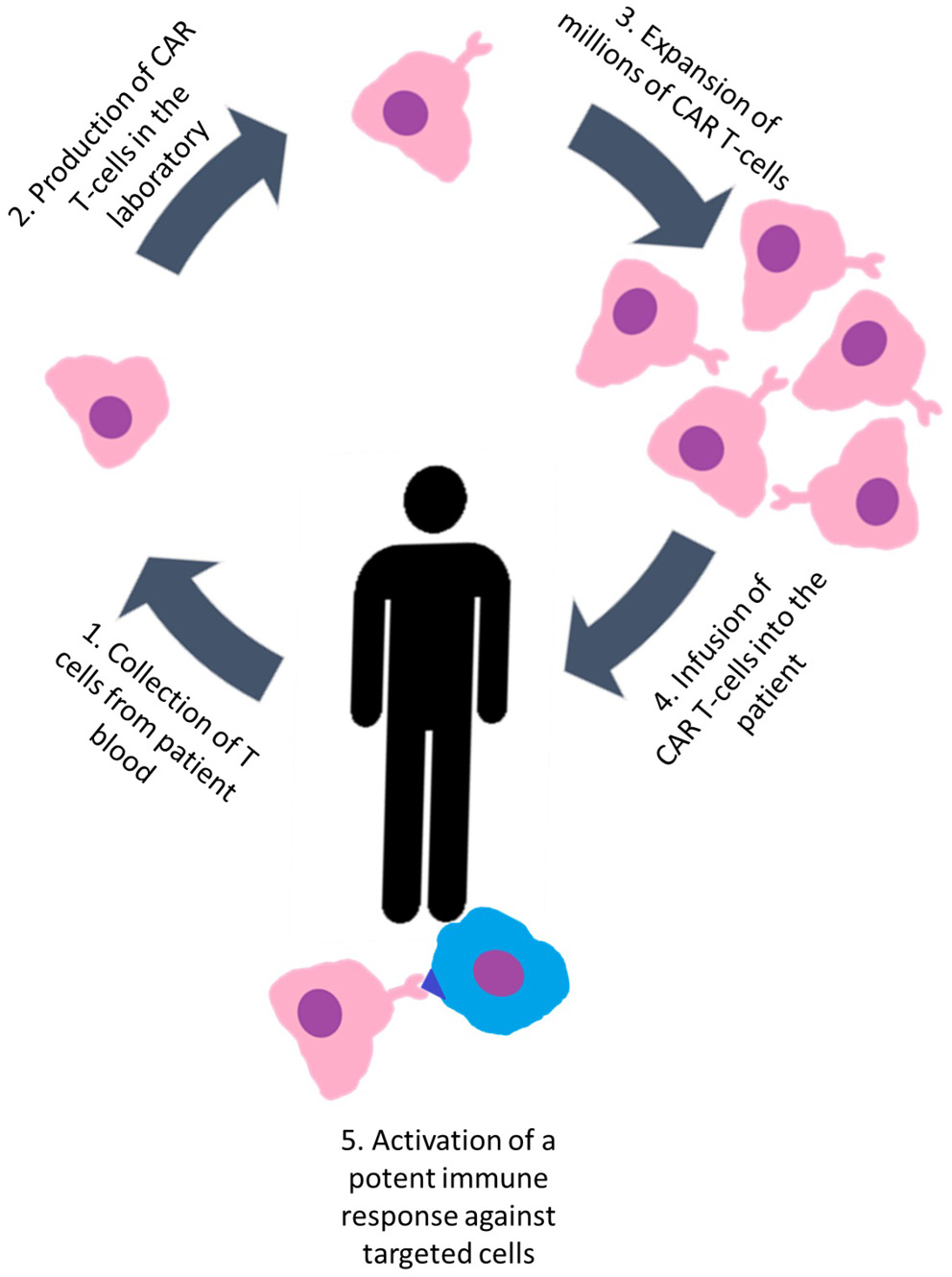
The CAR-T therapy involves taking a patient’s T-cells, which are genetically engineered to express chimeric antigen receptors (CARs) that target specific antigens, and then reintroducing these engineered cells into the patient. The result is the activation of a potent immune response against targeted cells (Figure 2).102 Recent scientific studies have reported on the application of anti-CD19 CAR-T cell-based therapy in individuals with treatment-resistant SLE. Preclinical studies in a mouse model have shown that therapeutic intervention in SLE with anti-CD19 CAR-T cells results in a reduction in the B-lymphocyte population, cessation of autoantibody formation and reversal of organ-related symptoms.103 Mougiakakos et al. reported the case of a 20-year-old patient with refractory SLE complicated by active nephritis who underwent the therapeutic intervention described above. After the administration of CAR-T therapy, the patient exhibited a rapid reduction in dsDNA autoantibodies and achieved clinical remission.104 Building on previous publications, the study by Mackensen et al. reports the results of 5 refractory SLE patients receiving CAR-T therapy. The results revealed a significant reduction in B-cell counts, normalization of clinical parameters and improvement in laboratory results, including a reduction in anti-dsDNA and anti-Sm antibody below detectable levels. Following a 3-month period, all enrolled patients demonstrated sustained SLE remission. The intervention’s safety profile shows a positive trend, with only mild cytokine release syndrome observed in some treated patients. However, larger placebo-controlled trials are needed to obtain comprehensive follow-up data.105 In another case study, a patient with a long (20 years) history of SLE complicated by stage IV diffuse large B-cell lymphoma was treated with a CAR-T construct expressing both anti-BCMA and anti-CD19. After an extended post-treatment period, sustained plasma cell depletion and durable remission were consistently observed, accompanied by undetectable anti-nuclear antibody titers.106 Chimeric antigen receptor cells offer a pivotal breakthrough in SLE therapy. However, further preclinical investigations and clinical trials are necessary to fully evaluate their potential.
Hematopoietic stem cells
Research endeavors targeting the treatment of SLE frequently incorporate stem cell transplantation, with a particular focus on hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs). The potential of hematopoietic stem cells transplantation (HSCT) as a therapeutic option for patients with SLE has been the subject of investigation over the past 2 decades. The cited literature review has identified limitations to this therapeutic approach that prevent its widespread clinical use. These constraints include the possibility of adverse effects, a significant tendency for relapse and higher financial burdens compared to biologic medications.107, 108
Scientific studies have identified changes in the characteristics of MSCs in individuals with SLE. Mesenchymal stem cells derived from SLE patients exhibit deficiencies, including aberrant cytokine secretion, compromised phenotypic features, diminished proliferation, and impaired immunomodulatory capacities.109 The therapeutic effectiveness of allogeneic mesenchymal stem cell therapy (MSCT) depends mainly on its systemic immunoregulatory effect on various immune regulatory cell populations, including T cells, B cells, plasma cells, dendritic cells, macrophages, and others.110 In addition, MSCs secrete a range of anti-inflammatory cytokines, which act as mediators in regulating immune responses. In addition, MSCs have the capacity to localize in kidney, lung, liver, and spleen tissues, where they may play a role in the regulation of local inflammatory processes.111 Over half of the patients with SLE experienced complete and partial clinical remission following MSCT. Mesenchymal stem cell therapy has been shown to induce remission in multi-organ dysfunction, such as LN. It is worth noting that mild side effects, such as dizziness and a feeling of warmth, were experienced by only a small number of patients.112 Nevertheless, it is imperative to emphasize the need for additional evidence from large clinical trials to validate the results observed in preclinical studies, while fully elucidating the therapeutic mechanisms underlying MSC treatment.
Limitations
It is important to acknowledge that cited studies and the therapies they propose have their limitations. The heterogeneity of SLE presents challenges at the trial design stage. Achieving a homogenous population is exceptionally difficult. Furthermore, the issue also lies in the small number of patients participating in the reviewed studies. Many scales are used to assess disease activity and treatment response, but there is often a lack of consistency between them. In addition, it is difficult to compare results between studies because the clinical endpoints of the studies are different. Finally, experimental therapies, especially HSC transplantation, carry a high risk of adverse events for a patient, which can lead to serious complications such as serious infections or even death.
Conclusions
In this review, we summarized recent achievements in the treatment of SLE. It is a heterogeneous disease, with a complex pathogenesis and an unpredictable course. Therefore, it is important to develop novel treatment modalities that address these challenges. For patients with suboptimal disease management, the inclusion of anifrolumab and voclosporin in treatment guidelines opens up new opportunities. Anifrolumab is the first biological drug that modulates an interferon signaling pathway that plays a major role in the SLE pathogenesis. At the moment, its results are noninferior to other recommended biologics. On the other hand, voclosporin is the first calcineurin inhibitor specifically indicated in the treatment of LN.
Other biological medications described in our review remain in phase II and III clinical trials, but they have already shown some promising potential. Plasma depletion therapy, CAR-T and HSCT remain an experimental therapy in SLE. Further research is needed to assess the safety and efficacy of the proposed treatment, as well as long-term results and side effects.