Abstract
Background. During ischemic stroke treatment, cerebral ischemia/reperfusion (I/R) injury results in neuronal cell death and neurological dysfunctions in brain. Previous studies indicate that basic helix-loop-helix family member e40 (BHLHE40) exerts protective effects on the pathology of neurogenic diseases. However, the protective function of BHLHE40 in I/R is unclear.
Objectives. This study aimed to explore the expression, role and potential mechanism of BHLHE40 after ischemia.
Materials and methods. We established models of I/R injury in rats and of oxygen-glucose deprivation/reoxygenation (OGD/R) in primary hippocampal neurons. Nissl and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed to detect neuronal injury and apoptosis. Immunofluorescence was used to detect BHLHE40 expression. Cell viability and cell damage measurements were conducted using Cell Counting Kit-8 (CCK-8) assay and lactate dehydrogenase (LDH) assay. The regulation of BHLHE40 to pleckstrin homology-like domain family A, member 1 (PHLDA1) was assessed using the dual-luciferase assay and chromatin immunoprecipitation (ChIP) assay.
Results. Cerebral I/R rats exhibited severe neuronal loss and apoptosis in hippocampal cornu Ammonis 1 (CA1) region, accompanied by downregulated BHLHE40 expression at both mRNA and protein levels, indicating that BHLHE40 may regulate the apoptosis of hippocampal neurons. The function of BHLHE40 in neuronal apoptosis during cerebral I/R was further explored by establishing an OGD/R model in vitro. Low expression of BHLHE40 was also observed in neurons treated with OGD/R. The OGD/R administration inhibited cell viability and enhanced cell apoptosis in hippocampal neurons, whereas BHLHE40 overexpression reversed those changes. Mechanistically, we demonstrated that BHLHE40 could repress PHLDA1 transcription by binding to PHLDA1 promoter. The PHLDA1 is a facilitator of neuronal damage in brain I/R injury and its upregulation reversed the effects caused by BHLHE40 overexpression in vitro.
Conclusions. The transcription factor BHLHE40 may protect against brain I/R injury through repressing cell damage via regulating PHLDA1 transcription. Thus, BHLHE40 may be a candidate gene for further study of molecular or therapeutic targets for I/R.
Key words: BHLHE40, PHLDA1, apoptosis, cerebral I/R injury, OGD/R
Background
Ischemic stroke (IS) is a serious disease endangering human health, which can lead to a disturbance in the local blood supply, eventually resulting in severe morbidity and mortality.1, 2 Angioplasty/stenting, mechanical thrombectomy and pharmacological thrombolysis with tissue membrane activator have been confirmed to be effective interventions for IS.3, 4 However, most IS patients still suffer from a poor prognosis due to severe neurological dysfunction. Cerebral ischemia/reperfusion (I/R) injury is one of the most prevalent complications of IS, which often occurs during thrombolysis or surgery.5 At present, I/R injury is a global health issue, necessitating more research into molecular processes as well as the identification of effective treatment targets.
Previous studies suggest that various neurological illnesses can lead to cognitive dysfunction6 and neurogenic inflammation.7, 8 After I/R, the physiological functions in numerous organs become more vulnerable to oxidative stress, cellular damage and death.9, 10 The process of programmed cell death (PCD) is essential in the development of the nervous system, and dysregulation of cell-death programs also occurs in ischemic disorders.11 There is mounting evidence that cerebral I/R can activate a variety of cell death pathways, including necrosis, apoptosis and autophagy-associated PCD.11, 12, 13 One of the best-characterized types of PCD is cell apoptosis, which is considered to be the underlying cause of I/R injury.11 Necroptosis and apoptosis as the 2 regulated cell death mechanisms are involved in mediating the neurologic damage in IS.14, 15 A systematic literature review concluded that the designated hindrance of apoptosis might be a compelling treatment for different neurodegenerative infections.16 According to reports, necrostatin-1 played a therapeutic role in ischemic brain injury as a small-molecule inhibitor of necroptosis.15 Thus, targeted inhibition of pro-apoptotic factors will give a viable remedial methodology to cerebral I/R injury.
BHLHE40, also named DEC1, STRA13 or SHARP2, is a member of the basic helix-loop-helix transcription factor family, and it is reported to involve various biological processes, such as the response to hypoxia, cell differentiation, inflammatory response, and tumorigenesis.17, 18, 19 It has been linked to a variety of biological processes in cell lines, including the control of the cell cycle and cell death and differentiation.20 The anti-apoptotic role of BHLHE40 was reported in many papers. There is evidence that BHLHE40 is abundantly expressed in different cancers and safeguards against apoptosis by inducing survivin, an anti-apoptotic protein.21 In colon carcinoma, BHLHE40 is highly expressed and plays an antagonizing role in serum deprivation-induced apoptosis, along with selectively inhibiting procaspase activation.22 Nevertheless, it is hazy whether BHLHE40 has a role also in neuronal apoptosis brought about by I/R injury. BHLHE40 is expressed in different tissues and acts as a transcriptional repressor or activator to repress or activate the transcriptional regulation of downstream target genes. For instance, BHLHE40 plays an important role in controlling the mammalian circadian rhythm by inhibiting the CLOCK/BMAL1-actuated promoter.23 BHLHE40 was able to increase the expression of TAp73 via transcriptional activation of the TAp73 promoter.24 In addition, BHLHE40 increases the promoter activation of T-box transcription factor Tbx21-mediated Ifng.25
Pleckstrin homology-like domain family A, member 1 (PHLDA1), also known as PHRIP, TDAG51 and DT1P1B11, exerts an important role in different pathologic states.26 The importance of PHLDA1 in the process of apoptosis has been reported in a variety of cancers.27, 28 In addition, it has been indicated that PHLDA1 inhibition ameliorates I/R induced injury, including myocardial injury and hepatic injury.29, 30 In addition, the protective role of PHLDA1 knockdown has been described in OGD/R-injured neurons, revealing that PHLDA1 may act as a target for neuroprotective therapy. Based on these findings, we inferred that PHLDA1 might exert a central role in the nervous system.
Objectives
Bioinformatics predicted that BHLHE40 could bind to PHLDA1. On this basis, the study aimed to investigate whether PHLDA1 could serve as a transcriptional target of BHLHE40, and to uncover the protective effects of BHLHE40 on I/R injury. Moreover, the function of PHLDA1 on cell apoptosis was explored in oxygen-glucose deprivation/reoxygenation (OGD/R)-triggered primary hippocampal neurons with overexpressing PHLDA1 and BHLHE40 simultaneously.
Materials and methods
Bioinformatics analysis
JASPAR is an open-access database storing manually curated transcription factors binding profiles as position frequency matrices.31 The binding between the transcription factor BHLHE40 and PHLDA1 promoter was predicted using the JASPAR website (http://jaspar.genereg.net/).
Animals
Male rats (Sprague Dawley, 6–8-week-old) were randomly allocated in the sham group and the I/R group. The rats were housed in a 12-hour light/dark cycle, with 45–55% humidity and temperature of 22 ±1°C, in controlled conditions, with food and water available freely. The 4-vessel occlusion (4-VO) method was used to induce transient cerebral ischemia. After 1 week of adaptive feeding, electrocautery was used to occlude both vertebral arteries after anesthesia with pentobarbital sodium (50 mg/kg). On the next day, both carotid arteries were ligated using microvascular clips for 10 min to instigate cerebral ischemia. After that, the aneurysm clips were removed for reperfusion and the models were established after 72 h of reperfusion. Then, the rats were sacrificed and the cerebrum tissues were isolated for subsequent investigations. Except that the bilateral common carotid arteries were not occluded, other operations in the sham group were the same as those in the I/R group. All animal procedures were approved by the committee of Jinan Central Hospital, Shandong University (approval No. JNCH2021-6).
Cell culture and lentivirus production
Hippocampal neurons obtained from the brains of newborn rats were dissociated using 0.125% trypsin (Sigma-Aldrich, St. Louis, USA) at 37°C for 15 min. Then, the cells were seeded in plates coated with 0.1 mg/mL of Poly-L-lysine (Solarbio, Beijing, China). The cells were then grown in a neurobasal medium (Gibco, Waltham, USA) containing 2% B-27 and 1% glutamine for 24 h at 37°C in a 5% CO2 environment. Subsequently, the medium was changed every 3.5 days.
The cells were cultivated in 6-well plates and incubated at 37°C and 5% CO2 overnight. After that, lentivirus overexpressing BHLHE40 (LV-BHLHE40) vector or containing the control vector (LV-vector) was used to infect hippocampal neurons. Additionally, lentivirus overexpressing PHLDA1 (LV-PHLDA1) vector or its control vector (LV-vector) was used to infect the hippocampal cells. To verify the regulation of BHLHE40 on PHLDA1, the cells were co-infected with LV-BHLHE40 and LV-PHLDA1.
OGD/R application
After a 72-hour lentivirus infection, OGD/R was initiated. Briefly, Dulbecco’s modified Eagle’s medium (DMEM) was used to replenish neurons and then the cells were placed in an anaerobic chamber filled with 85% nitrogen, 10% hydrogen and 5% CO2 at 37°C for 4.5 h. A fresh neurobasal medium was supplanted after OGD treatment, and the neurons were then incubated at 37°C/5% CO2 for 24 h.
Brain water content measurement
The brain was removed from 4-VO-conducted rats, and the wet weight of the brain was measured immediately. After drying in a vacuum oven at 121°C for 24 h, the brains were re-weighed for the dry weight. Finally, the degree of brain edema was calculated as follows: (wet weight – dry weight)/wet weight × 100%.
Nissl staining
Nissl staining was assessed to detect neuronal injury in the cornu Ammonis 1 (CA1) and CA3 regions of the rat hippocampus. Sections were stained with 0.5% cresyl violet for 10 min and differentiated in 0.25% acetic acid in ethanol for a few seconds. Using an optical microscope (model DP73; Olympus Corp., Tokyo, Japan), we captured the pictures at a magnification of ×40 and ×200. The Nissl-positive cells were counted using the ImageJ software (National Institutes of Health, Bethesda, USA), and the quantitative data were expressed as the percent of Nissl-positive cells (%).
TUNEL staining
The sections of neuronal cells or paraffin-embedded brain sections were permeabilized using 0.1% Triton X-100 (Beyotime Biotechnology, Shanghai, China). After that, the sections were stained with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) fluorescence staining reagents at 37°C in the dark for 60 min, and then stained with DAPI (4′,6-diamidino-2-phenylindole; Aladdin, Shanghai, China). Fluorescent images were captured using an optical microscope (model DP73; Olympus Corp.). Quantitative data were expressed as the percent of TUNEL-positive cells (%).
Quantitative real-time polymerase
chain reaction (qRT-PCR)
Expressions of BHLHE40 and PHLDA1 mRNA were quantitatively determined with quantitative real-time polymerase chain reaction (qRT-PCR). TRIpure reagent (BioTeke, Wuxi, China) and BeyoRTTM II M-MLV reverse transcriptase (Beyotime Biotechnology) were used to isolate total RNA and synthesize cDNA respectively. For the conduction of qRT-PCR, SYBR Green (Solarbio) was used, and the data obtained from the system was determined by calculating the 2−ΔΔCT relative fold change. The mRNA levels of BHLHE40 and PHLDA1 were normalized to β-actin. The primer sequences were as follows (5’-3’): BHLHE40 F: AGCGAGGACAGCAAGGA; BHLHE40 R: CCAAGTGACCCAAAGTAGTAAG; PHLDA1 F: ACAGCCGAACCGTCCCA; PHLDA1 R: TTTGCCCTCCGCCATCA.
Western blot
Whole-cell lysates were collected in the lysis buffer and the BCA method (Beyotime Biotechnology) was used to determine the protein concentration. Then, we subjected equal amounts of protein to sodium dodecyl sulfate-polyacrylamide gel electrophoresis) (SDS-PAGE) and transferred them to polyvinylidene difluoride (PVDF) membranes. Subsequently, the membrane was incubated with specific primary antibodies (BHLHE40 (Abclonal, Wuhan, China); PHLDA1 (Affinity Biosciences, Changzhou, China); caspase-3 (Cell Signaling Technology (CST), Danvers, USA); cleaved caspase-3 (CST); Bcl-2 (Abclonal); PARP (CST); cleaved PARP (CST); BAX (Affinity); and β-actin (Beyotime Biotechnology)) at 4°C overnight and subsequently incubated with secondary antibodies (Beyotime Biotechnology) at 37°C for 1 h. The quantitative densitometric values of the proteins were normalized to that of β-actin.
Immunofluorescence
For double immunofluorescence, paraffin-embedded hippocampus sections (5 μm) were incubated at 60°C in an oven for 2 h, followed by dewaxing and dehydrating. The sections were incubated with primary against BHLHE40 (Rabbit; Abclonal) and NeuN (Mouse; Abcam, Cambridge, UK) antibodies at 4°C overnight. Then, the sections were incubated with the secondary antibody conjugated to fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit immunoglobulin G (IgG; Abcam) and cyanine 3 (Cy3)-labeled goat anti-mouse IgG (Invitrogen, Carlsbad, USA) at 37°C for 1 h. Phosphate-buffered saline (PBS) containing 0.1% Triton X-100 was used to permeabilize the fixed cells for 30 min at room temperature. The cells were incubated successively with the BHLHE40 antibody and the secondary antibody against FITC-labeled goat IgG. The number of BHLHE40-positive cells and BHLHE40/NeuN double-positive cells were counted at ×400 magnification using ImageJ software.
CCK-8
A CCK-8 Cell Proliferation Detection Kit (KeyGen Biotech, Nanjing, China) was used to determine cell viability. Briefly, CCK-8 solution (10 μL) was added and incubated at 37°C for 2 h with 3×103 cells seeded into 96-well culture plates. A microplate reader (Biotek, Winooski, USA) was used to measure the optical density (OD) at 450 nm.
Lactate dehydrogenase assay
The lactate dehydrogenase (LDH) assay kit (Jiancheng Bioengineering Institute, Nanjing, China) was used to detect cell damage. The LDH activity was determined by measuring the absorbance at 450 nm.
Dual-luciferase assay
To generate PHLDA1 promoter-luciferase construct, a fragment of PHLDA1 promoter from –1956 bp to +12 bp was amplified with PCR and cloned to pGL3 vectors. Then the pGL3 vectors were transfected with BHLHE40 overexpression (OE-BHLHE40) or control vectors (OE-vector) into the hippocampal cells using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instruction. An assay kit (KeyGen Biotech) for determining luciferase activity was utilized in cells 48 h after transfection.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was carried out using a cell chip kit (Wanleibio, Shenyang, China) according to the manufacturer’s instructions. In brief, neurons were cross-linked to fix the DNA/protein complexes containing 1% formaldehyde for 10 min at ambient temperature. Glycine was added at a final concentration of 0.125 M to terminate the reaction. At 4°C, cell lysate was sonicated for 2 min, and the samples were mixed with precleared protein A/G beads for 2 h. Then, the supernatant fraction was incubated with the BHLHE40 antibody and normal IgG overnight at 4°C. After releasing the crosslink, purified DNA was subjected to PCR and analyzed using agarose gel electrophoresis. The primer sequences were as follows (5’-3’): chip PHLDA1 F: GGGGAAAGGGAATAACA; chip PHLDA1 R: GTCTCGGTCAAACAAGG.
Statistical analyses
Statistical significance was determined using GraphPad Prism v. 8.0 (GraphPad Software, San Diego, USA). The normality of distribution of all data was checked with D’Agostino–Pearson omnibus test. A value of p < 0.05 was deemed significant.
For data in 2 groups, the homogeneity of variance was examined with F test. If data complied with the normal distribution and had equal variance, p-values were calculated using unpaired t-test; if not, p-values were determined using Mann–Whitney U test.
For data from multiple groups, the homogeneity of variance was examined using Brown–Forsythe test. If data were in normal distribution and had unequal variances, p-values were calculated with one-way analysis of variance (ANOVA) followed by Tukey’s post-test; if not, p-values were determined using Kruskal–Wallis test followed by Dunn’s post-test.
Results
BHLHE40 is downregulated in rats undergoing cerebral I/R injury
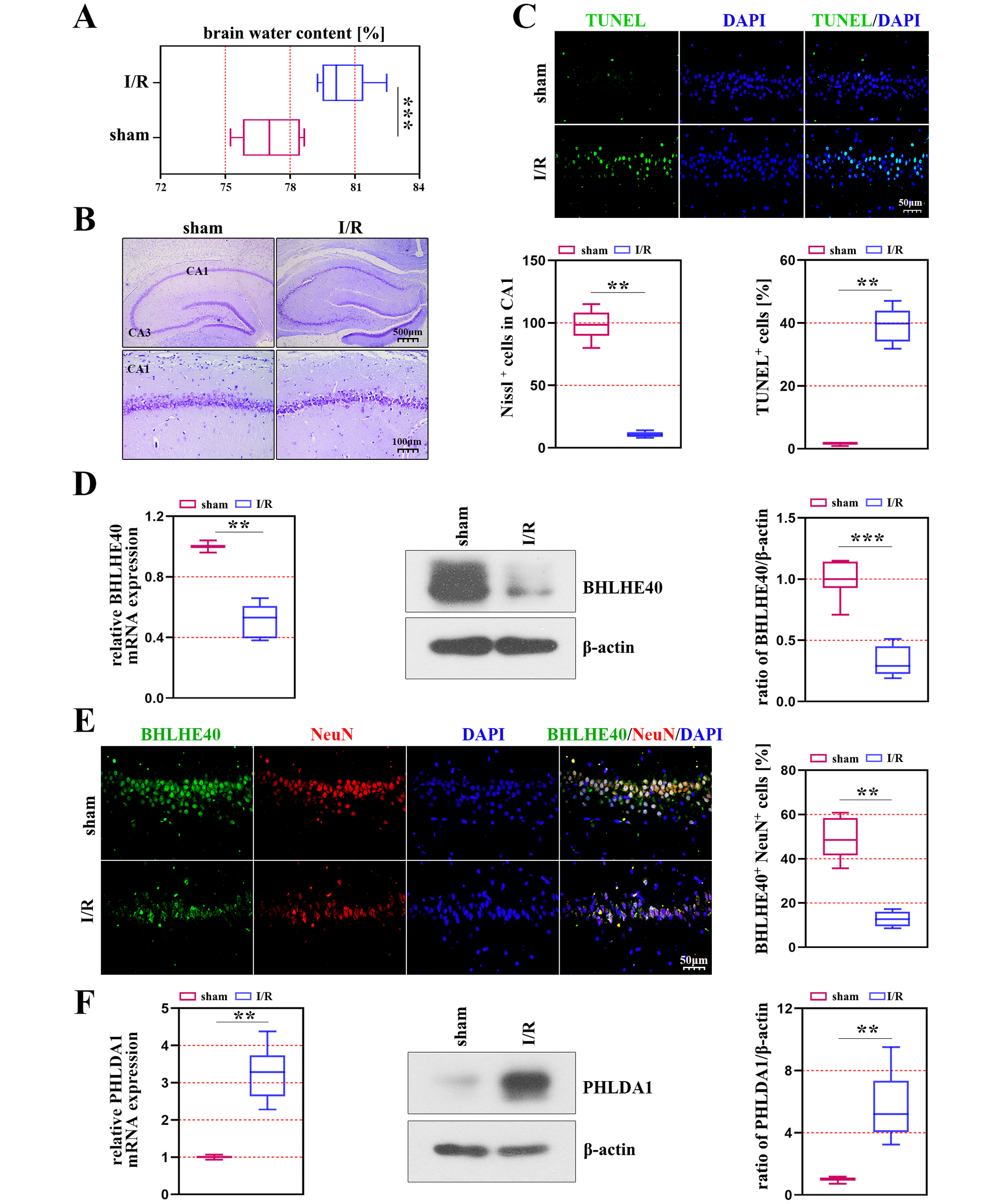
Our study investigated the role of BHLHE40 by creating a model of brain I/R injury on rats, and we discovered that the brain water content was significantly increased in the ischemic rats compared with the control rats (Figure 1A). The results of Nissl staining indicated that the neurons were changed after ischemia, which presented neuronal cell loss and karyopyknosis in the CA1 region of rats (Figure 1B). The number of apoptotic cells was increased in the ischemic models compared with the sham group (p < 0.001, Figure 1C). We found that the mRNA and protein levels of BHLHE40 had a notable decrease in rats induced with induced I/R (Figure 1D). The results of immunofluorescence staining suggested that BHLHE40 co-localized with NeuN (a neuronal marker), and positive cells of BHLHE40 and NeuN were decreased in the CA1 region of the hippocampus after I/R injury (p < 0.001, Figure 1E). As shown in Figure 1F, the transcriptional and protein levels of PHLDA1 were markedly increased in rats induced with induced I/R.
BHLHE40 overexpression ameliorates cell damage in OGD/R-induced neurons
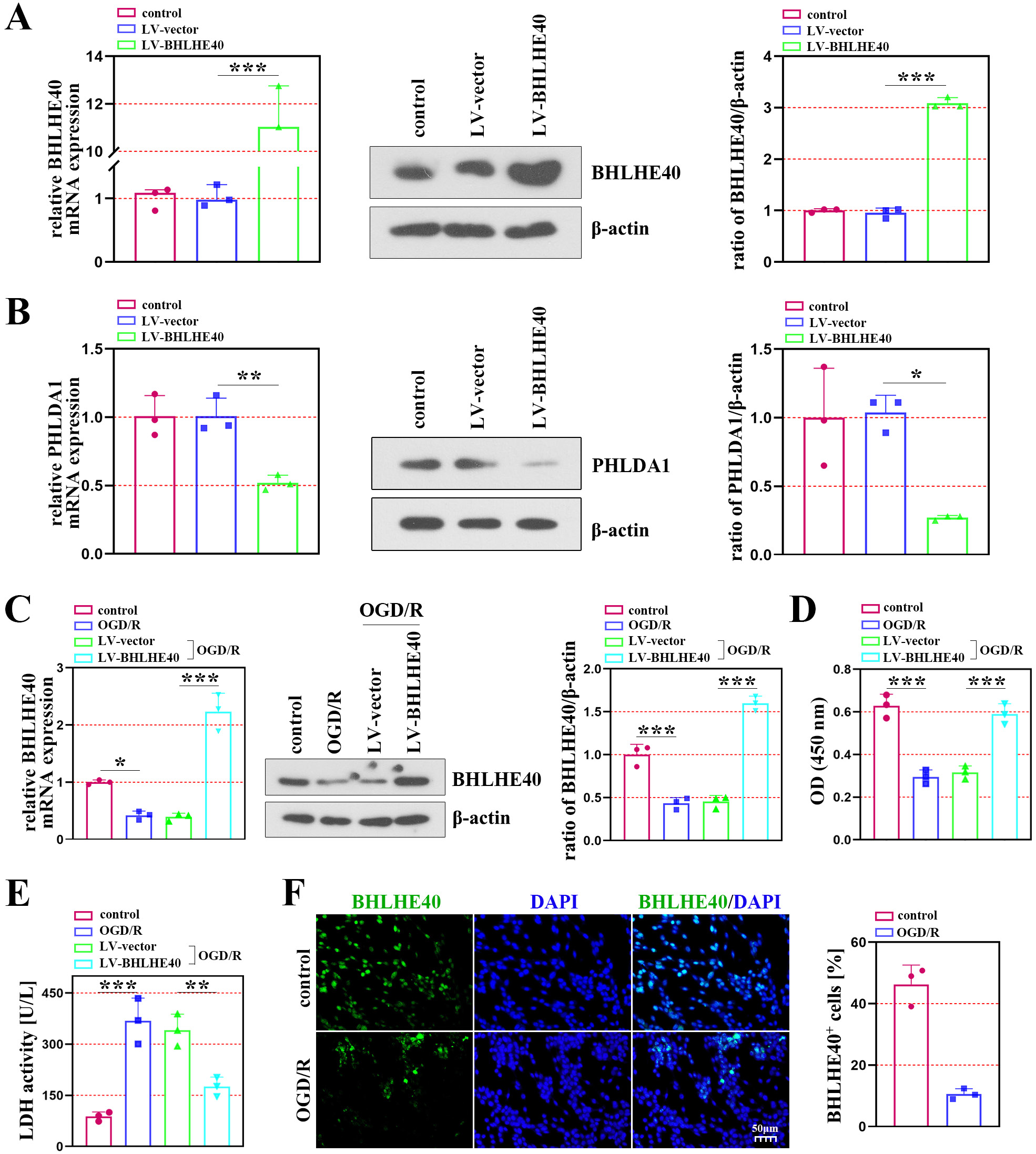
Using neuronal cells subjected to OGD/R, we analyzed cell damage and apoptosis to determine the role of BHLHE40 in brain I/R injury. The efficiency of lentivirus infection was verified using qRT-PCR and western blot assays (Figure 2A). The results showed that the mRNA and protein levels of PHLDA1 were decreased after BHLHE40 overexpression (Figure 2B). Figure 2C shows that OGD/R treatment downregulated BHLHE40 expression, while BHLHE40 overexpression significantly elevated its expression in OGD/R-induced neurons (Figure 2C). As shown in Figure 2D,E, the OGD/R-induced cells activated the cell viability and inhibited the LDH activity after BHLHE40 overexpression. Immunofluorescence staining showed decreased BHLHE40-positive cells in the OGD/R group (Figure 2F).
BHLHE40 overexpression ameliorates apoptosis in OGD/R-induced neurons
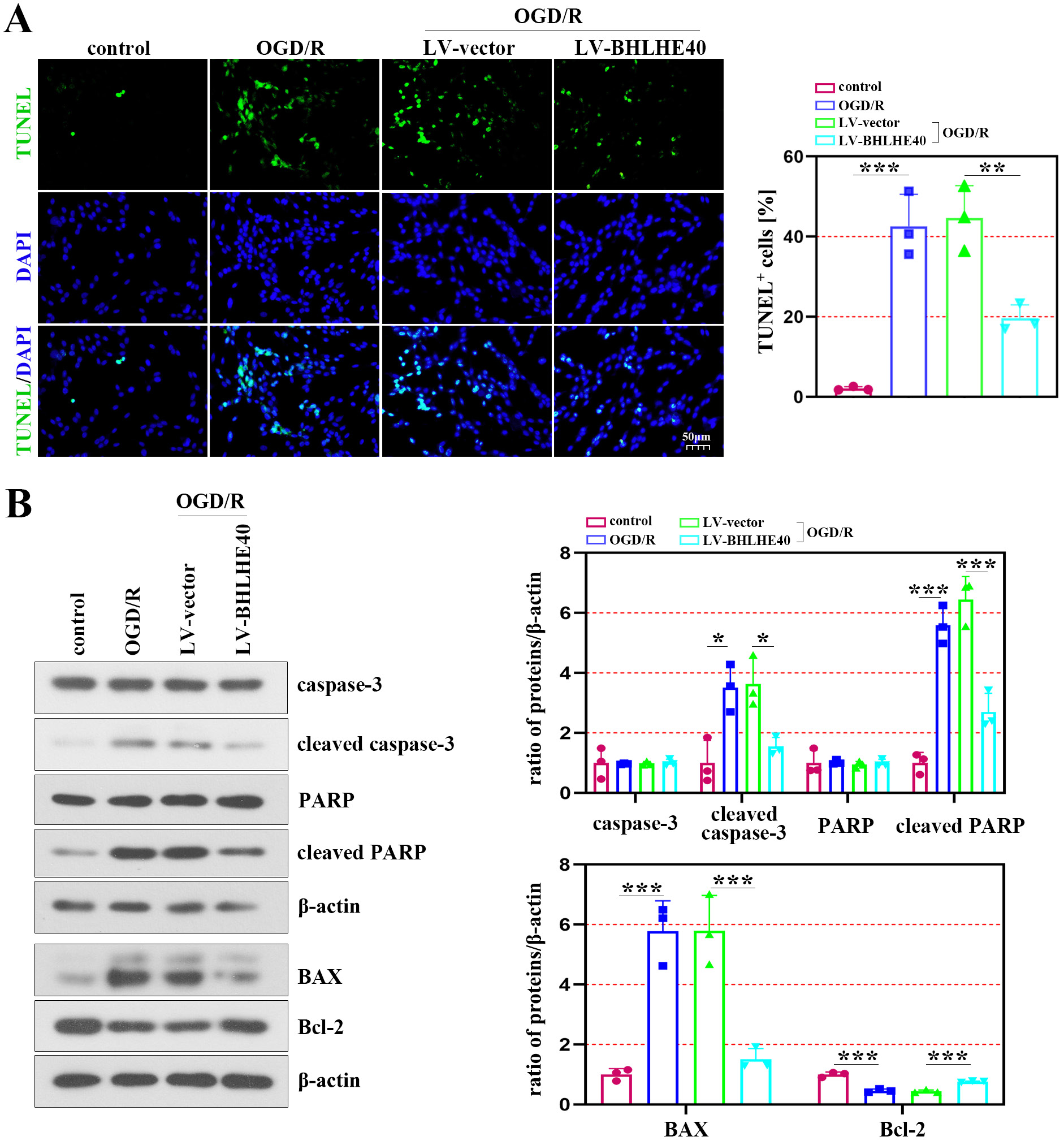
The ratio of TUNEL-positive cells was significantly increased after OGD/R administration, whereas BHLHE40 overexpression inhibited the effects (Figure 3A). The western blot analysis indicated that the protein levels of cleaved caspase-3 and cleaved PARP were markedly decreased in OGD/R-induced cells with BHLHE40 overexpression, while the levels of caspase-3 and PARP did not change (Figure 3B – upper section). In addition, the results showed that BAX was downregulated in the presence of BHLHE40 overexpression, which enhanced the protein content of Bcl-2 (Figure 3B – lower section).
Transcriptional regulatory effects of BHLHE40 on PHLDA1
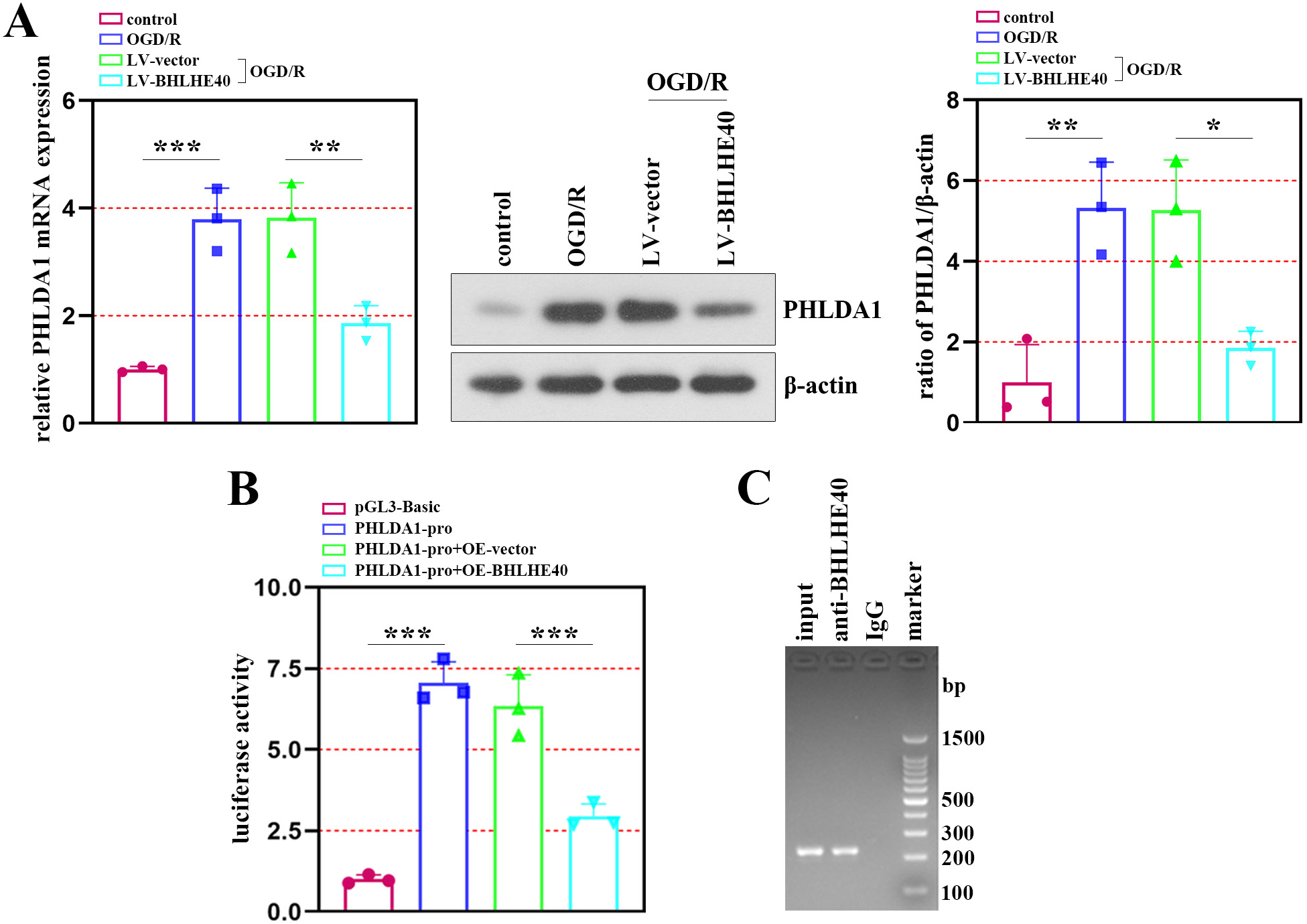
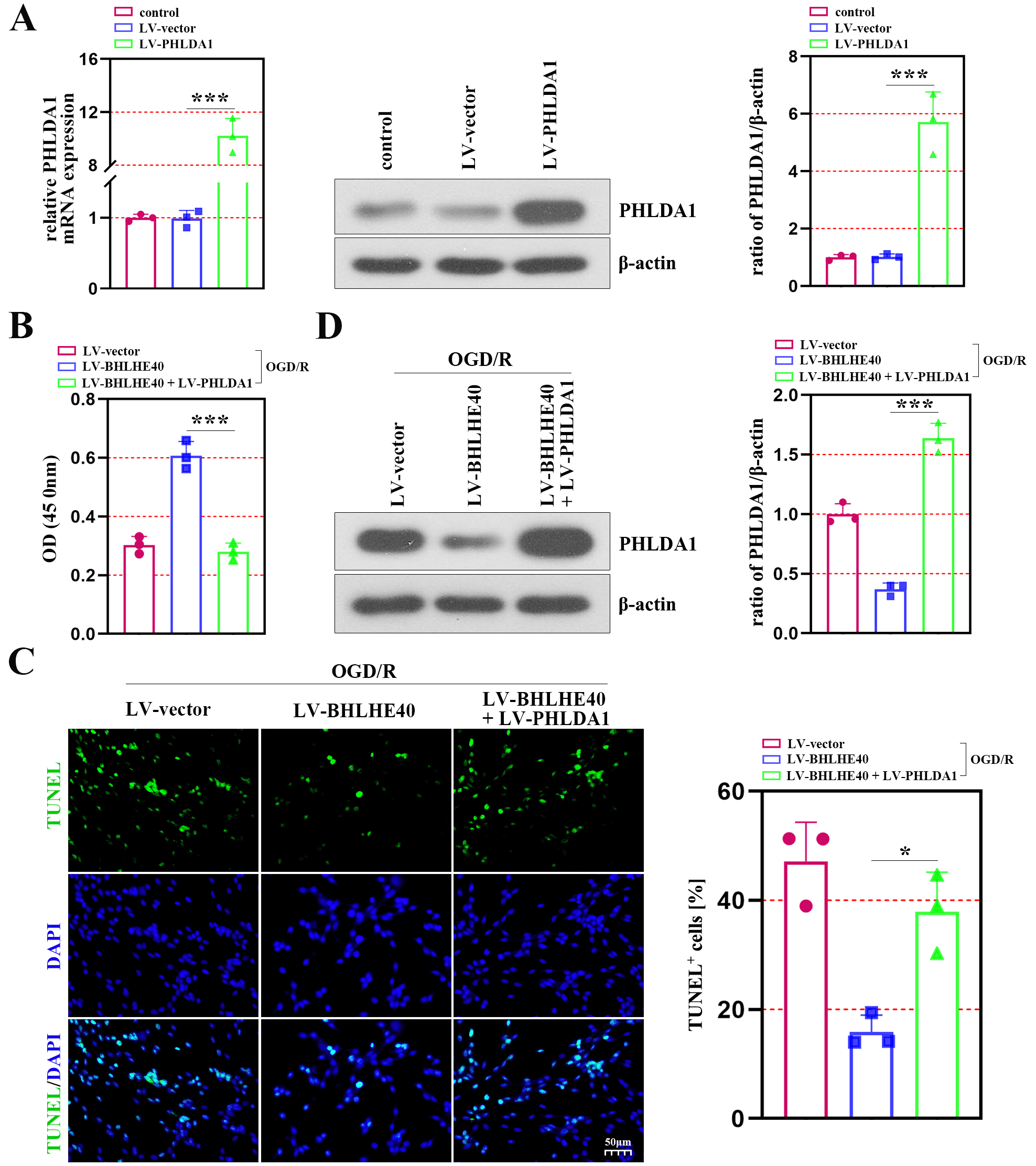
To clarify the association between BHLHE40 and PHLDA1 in neuronal injury after OGD/R treatment, we identified PHLDA1 expression after BHLHE40 upregulation. The results of qRT-PCR and western blot assays showed that PHLDA1 expression was negatively regulated by BHLHE40 (Figure 4A). Then, a luciferase assay was performed using a plasmid constructed with the PHLDA1 promoter linked to a luciferase reporter gene, and it showed that BHLHE40 repressed the luciferase activity of the PHLDA1 promoter (Figure 4B). Furthermore, ChIP experiments demonstrated that PHLDA1 was the target gene of BHLHE40 (Figure 4C).
BHLHE40 overexpression inhibits cell apoptosis of OGD/R-induced cells through controlling PHLDA1
To verify the mechanism of BHLHE40 through regulating PHLDA1 in I/R injury, the hippocampal neurons were co-infected with LV-BHLHE40 and LV-PHLDA1, and subjected to OGD/R treatment. The mRNA and protein levels of PHLDA1 verified the efficiency of lentivirus infection (Figure 5A). As shown in Figure 5B, the enhanced cell viability caused by BHLHE40 upregulation was inhibited in the presence of PHLDA1 overexpression. The anti-apoptotic role of BHLHE40 in OGD/R-induced neurons was observed using the TUNEL staining, whereas the ratio of TUNEL-positive cells was increased after PHLDA1 overexpression (Figure 5C). We discovered that the inhibiting effect of BHLHE40 overexpression on PHLDA1 expression was reversed by PHLDA1 upregulation in neurons subjected to OGD/R (Figure 5D).
Discussion
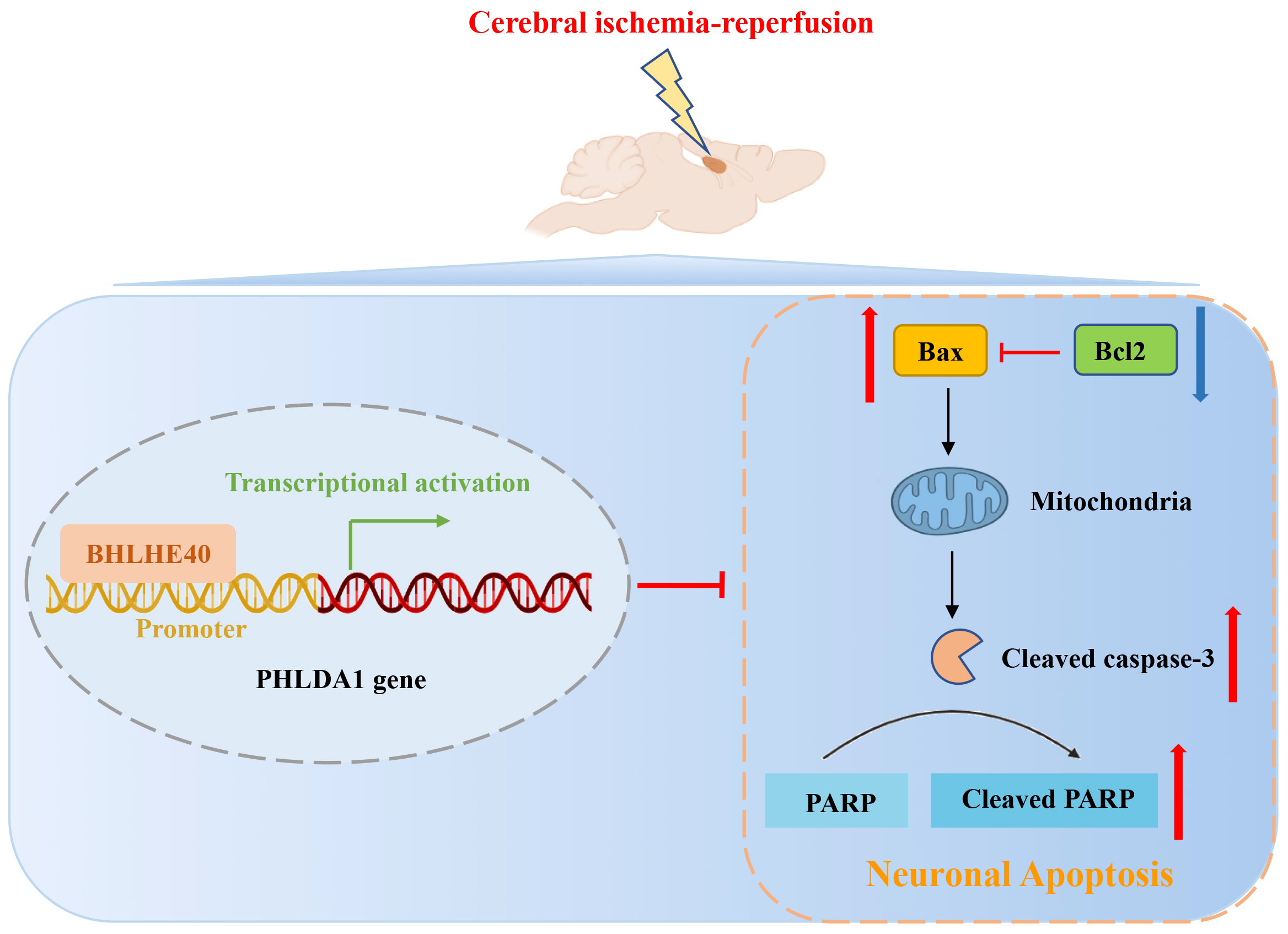
This study found that BHLHE40 was downregulated in ischemia rats and primary neurons following OGD/R treatment. To systematically identify the function of BHLHE40 in brain I/R injury, cell damage and apoptosis were measured in BHLHE40-overexpressed neurons. In vitro experiments further confirmed the direct transcriptional regulation of PHLDA1 by BHLHE40. Furthermore, BHLHE40 overexpression shielded neurons from damage from brain I/R via inhibiting PHLDA1.
It is well known that neuronal apoptosis can shape the developing brain,16 and dysregulation of apoptosis is reported to be related to the progression of neurodegenerative illnesses and chronic diseases such as ischemic stroke.32 We found that increased ratio of TUNEL-labeled cells was observed in the hippocampal CA1 region of ischemia rats, and the neurons presented karyopyknosis. Several proteins are involved in mammalian apoptosis, including genes encoding Bcl-2, the adaptor protein Apaf-1, and cysteine protease caspases.16 A series of morphological and histochemical changes occur after apoptosis, and they are mainly the result of the activation of cellular suicide cysteine proteases – the caspases.33, 34 These enzymes subsequently participate in a cascade when pro-apoptotic signals are activated.33 Moreover, studies have indicated that the activity of Bcl-2 family gene products is involved in cellular and neurological processes in ischemic brain injury.35 Consistently, our study discovered that the expression of Bcl-2 was downregulated in ODG/R-induced neurons. In addition, BAX, a pro-apoptotic member of the Bcl-2 family, is required for neuronal death,36 and PARP-1 activity is suppressed by Bcl-2 directly.37 Previous study suggests that amantadine and topiramate can improve I/R injury by reducing apoptosis.38 We found that the protein accumulation of cleaved caspase-3, cleaved PARP and BAX was enhanced in ODG/R-induced neurons, while BHLHE40 overexpression reduced the protein amount, indicating that BHLHE40 exerted an anti-apoptotic role in I/R injury.
In addition to cellular damage, patients with brain damage present cognitive dysfunction39; thus, it is imperative to develop new techniques and therapeutic schedules. Accumulating evidence indicates that BHLHE40 exerts protective effects in different diseases. This was exemplified in the paper by Huynh et al., which reported that BHLHE40 was an essential repressor of cytokine IL-10 during Mycobacterium tuberculosis infection.40 In human hepatocellular carcinoma HepG2 cells, BHLHE40 upregulation partially antagonized apoptosis after 8-methoxypsoralen treatment and decreased the activation of caspase-3.41 Hamilton et al. found that mice lacking BHLHE40 displayed increased neuronal excitability and poorer synaptic plasticity in the hippocampal area.42 Similarly, our study confirmed the effects of BHLHE40 in neuroprotection, which was evidenced by the decreased protein amounts of apoptosis-related factors and cell apoptosis after BHLHE40 overexpression in neuronal cells with OGD/R treatment. In addition to the role of anti-apoptotic factor, BHLHE40 also ameliorated cell damage (according to the data from the detection of LDH activity). It was reported that BHLHE40 overexpression alleviated MPP+-induced neurotoxicity in Parkinson’s disease, whereas BHLHE40 silencing aggravated the symptom,43 which was consistent with our work.
The most critical function of BHLHE40 is as a transcription factor to regulate the expression of downstream target genes. A few molecules have been detailed to be included within the metastasis controlled transcriptionally by BHLHE40. Studies indicate that BHLHE40 plays a pro-survival and pro-metastatic role in breast cancer cells by activating the transcription of heparin-binding epidermal growth factor.17 Beyond that, a report by Kanda et al. suggested that BHLHE40 increased the promoter activation of T-box transcription factor Tbx21-mediated Ifng.25 Hereon, PHLDA1 was revealed to be repressed transcriptionally by BHLHE40, and functional investigation found that BHLHE40 overexpression inhibited cell apoptosis of OGD/R-induced cells via controlling PHLDA1. It is of pivotal importance to investigate the mechanism of BHLHE40 in ischemic injury.
PHLDA1, first identified by Park et al, was reported to fulfill an essential role in the induction of apoptosis in mouse T-cell hybridomas.44 In previous studies, PHLDA1 was markedly expressed in oxidative stress-induced cardiomyocyte and myocardial I/R wounds, and its upregulation in cardiomyocytes exacerbated oxidative stress-induced cardiomyocyte injury.29 In addition, other work has shown that PHLDA1 deficiency protects against OGD/R injury, which makes it one of the new targets for treating cerebral injury.45 Consistently, in the study, the data showed that PHLDA1 was upregulated in the hippocampal neurons exposed to OGD/R, and PHLDA1 overexpression inhibited the cell viability and promoted the cell apoptosis caused by BHLHE40 upregulation. Further studies showed that PHLDA1 was the target gene of BHLHE40, and BHLHE40 overexpression inhibited cell damage through controlling PHLDA1. The regulation between BHLHE40 and PHLDA1 might provide a novel direction for the development of neuroprotective therapy. The cardiac autonomic dynamics reported by Battaglia et al. provide a new understanding of the nervous system.46 There is growing evidence for the protective role of neuropsychiatric disorders,47, 48 which provides theoretical and clinical implications. Moreover, many new applications for psychiatric and neurological disorders have been reported, including non-invasive stimulation techniques49, 50 and prognostic biomarkers.51 Whether these applications can treat cerebral I/R injury remains to be explored in future studies.
Limitations
Some limitations of the present study should be recognized. First, although we provide potential therapeutic directions for clinical treatment, there is still no evidence to prove whether our results apply to humans. Moreover, the neurons were not treated with OGD/R under different conditions, so the optimal time for the establishment of the cell model was not obtained. The statistical analysis was exploratory, and the Bonferroni correction was not applied.
Even though the distribution of the data cannot be convincingly determined for very small samples, we assume that the observations come from the normal distribution. We agree that if this assumption is not true, the reported p-values and confidence intervals are unreliable and must be interpreted with caution. The nonparametric tests only indicated significant differences between groups in Figure 2A (BHLHE40 protein expression), Figure 2C (BHLHE40 mRNA expression) and 5A,B (PHLDA1 protein expression).
Conclusions
In summary, neuronal loss and apoptosis were found in the hippocampal CA1 region of cerebral I/R rats, and the results suggested that BHLHE40 and PHLDA1 were respectively downregulated and upregulated in rat brains after cerebral I/R. Functional analysis suggested that BHLHE40 suppressed neuronal apoptosis and cell death, as well as increased cell viability in OGD/R-induced cells. Mechanistic analysis reveals that BHLHE40 could decrease the expression of PHLDA1 through transcriptional inhibition of the PHLDA1 promoter, demonstrating that PHLDA1 serves as a transcriptional target of BHLHE40. These findings indicate that BHLHE40 protected against I/R damage by inhibiting PHLDA1. It is plausible that BHLHE40 may be an effective therapeutic option for cerebral I/R injury.