Abstract
A substantial increase in the interest in transthyretin cardiac amyloidosis (ATTR-CA) is a result of the constantly growing number of patients, the use of clear diagnostic protocols and the availability of the first selective drug for these patients. This has also raised the awareness of the disease among physicians of all specialties. The topic is particularly relevant to cardiologists, who use non-invasive multimodal imaging in their daily practice.
The differential diagnosis of the causes of myocardial hypertrophy includes arterial hypertension, hypertrophic cardiomyopathy, aortic stenosis (AS), athletic heart syndrome, Fabry disease, and cardiac amyloidosis (CA). It turns out that in patients with myocardial hypertrophy >15 mm, amyloidosis is the most common cause of left ventricular (LV) hypertrophy. In parallel, CA is one of the most common infiltrative diseases leading to a clinical picture that may mimic heart failure with preserved ejection fraction (HFpEF).
The accumulation of amyloid in the extracellular space impairs the diastolic function of the myocardium, which is observed as the restrictive cardiomyopathy phenotype. In advanced cases, the LV systolic function is also impaired. Moreover, protein deposits contribute to the disturbances of calcium metabolism and cell metabolism as well as to cardiotoxicity, leading to edema and damage to cardiomyocytes.
Key words: cardiomyopathy, amyloidosis, multimodal imaging, tafamidis, apical sparing
Introduction
The emergence of new therapies and guidelines for the management of patients with cardiac amyloidosis (CA) has increased the interest in this disease and brought a new approach to the diagnosis of the causes of myocardial hypertrophy.1, 2, 3 The position statement of the European Society of Cardiology (ESC) Working Group on Myocardial and Pericardial Diseases recommends screening for cardiac involvement if left ventricular (LV) hypertrophy exceeds 12 mm and coexists with specific red flags or a clinical scenario.3 An invasive diagnostic pathway, referring to all forms of CA, comprises solely cardiac biopsy positive for amyloid or a combination of an extracardiac biopsy positive for amyloid, and a presence of characteristic for CA findings on echocardiography or cardiac magnetic resonance. So far, there has been great awareness of storage diseases, particularly among echocardiographers, and the diagnosis of patients has often been limited to the exclusion or confirmation of light-chain cardiac amyloidosis (AL-CA), the treatment of which remains the domain of hematologists. If transthyretin cardiac amyloidosis (ATTR-CA) is suspected, a non-invasive diagnostic pathway is recommended, including scintigraphy, haematologic tests and echo/cardiac magnetic resonance (CMR).3
Cardiac amyloidosis manifests itself as a heart failure (HF) with initially preserved and – in more advanced stages – reduced ejection fraction, commonly with concomitant LV hypertrophy.4 Since amyloid deposits were found post-mortem in 25% of the unselected elderly population5 and in 13–19% of those with a history of HF with preserved ejection fraction (HFpEF),6 CA tends to be an underdiagnosed condition. Seferović et al., in a position paper from the Heart Failure Association of the ESC, draws attention to the possible change in the clinical phenotype of amyloidosis.7 In the natural history of the disease, depending on the timing of the diagnosis, the phenotype can be hypertrophic cardiomyopathy (frequently asymptomatic), restrictive cardiomyopathy (mildly symptomatic) or end-stage cardiomyopathy with severe LV contractile dysfunction and advanced HF symptoms.
The development of cardiological diagnostics enabled the non-invasive differentiation of the causes of myocardial hypertrophy, which proved to be particularly valuable in patients with HFpEF. This differential diagnosis includes arterial hypertension, hypertrophic cardiomyopathy, aortic stenosis (AS), athletic heart syndrome, Fabry disease, and CA. It turns out that in patients with myocardial hypertrophy >15 mm, amyloidosis is the most common cause of LV hypertrophy.8
Various heterogeneous pathophysiological processes are responsible for HFpEF, including a systemic inflammatory reaction, the dysfunction of large and small vessels as well as the fibrosis and remodeling of the affected tissues.9 There are clinical differences between amyloid cardiomyopathy and inflammatory metabolic HFpEF. Cardiomyopathy associated with ATTR-CA primarily affects elderly men with normal or low blood pressure, and the hypertrophy of the heart wall muscle is significant and affects all chambers. The LV filling pressure is high, the LV end-diastolic volume is reduced and a disproportionate increase in the natriuretic peptide concentrations is observed. On the other hand, HFpEF caused by other factors more frequently affects women, usually middle-aged, often with metabolic syndrome and other chronic inflammatory diseases or hormonal disorders. The LV walls are usually slightly thickened and the LV end-diastolic volume is normal or slightly increased. Other parameters that distinguish the remaining forms of HFpEF from CA are usually only slightly elevated natriuretic peptides, significantly increased systolic blood pressure (SBP) and increased inflammatory parameters.10
Transthyretin cardiac amyloidosis is also considered a common pathology accompanying AS (Figure 1).11, 12 The coexistence of CA with moderate or significant AS is estimated in up to 16% of patients over 80 years of age.13, 14 In several studies, the coexistence of amyloidosis and AS was assessed at 14–16% in elderly patients undergoing transcatheter aortic valve implantation (TAVI).13, 14, 15
The clinical and echocardiographic profile of patients with ATTR-CA and AS is quite similar. It seems that it particularly often affects elderly men with low-flow, low-gradient AS with preserved ejection fraction and reduced ejection volume.13 Chacko et al. showed that the coexistence of ATTR-CA and AS was associated with a poor prognosis and a significantly shorter survival time (22 months compared to 53 months; p = 0.001).16 These authors also noted statistically significantly longer survival for patients who underwent TAVI as compared to those who did not receive any treatment for severe AS (p = 0.012).16
Establishing the relationship and coexistence of AS and amyloidosis seems important, because an improvement after surgical valve replacement in these patients may be limited due to both, a higher risk of complications and death and the lack of clinical improvement caused by the presence of amyloid deposits in the heart. Therefore, it is recommended that the diagnosis of patients with a characteristic echocardiography image confirming the coexistence of significant LV hypertrophy and AS should be extended to include radioisotope testing, in order to exclude or confirm the presence of ATTR-CA.
Scully et al. showed that the TAVI procedure seemed to be promising for patients with ATTR-CA and AS.17 Periprocedual complications (p = 0.780) and deaths (p = 0.710) in the groups with and without amyloidosis did not differ statistically, and TAVI turned out to be more favorable than conservative treatment (p = 0.030).17 Recently published data showed that TAVI should not be delayed; post-TAVI survival in patients with CA and AS was no different than in lone AS.18
Pathogenesis – forms of amyloidosis
Light-chain cardiac amyloidosis is the most common form of CA, accounting for approx. 70–80% of all forms of the disease.19 What is characteristic of AL-CA are the deposits of monoclonal light chains that are produced by clonal bone marrow plasmacytes and, as a result of dissociation, accumulate in tissues and organs in the form of misfolded and insoluble proteins, called amyloids. The basis for the diagnosis of AL-CA is the detection of amyloid in a given organ or tissue during the histopathological examination (the presence of amyloid when staining the tissue material with Congo red), the assessment of the concentrations of kappa and lambda light chains and their ratio in serum free light chains (sFLCs) as well as the assessment of immunofixation (monoclonal protein) in blood serum and urine. The reference technique is the analysis of the amyloid composition using mass spectrometry, but this method is still not widely available.20 The use of chemotherapy or autologous hematopoietic stem cell transplantation (auto-HSCT) in the treatment of AL-CA depends, among other things, on how much various organs are affected. The Mayo Amyloidosis Staging System uses biomarkers to assess heart involvement.21 In untreated AL-CA, the survival is estimated at less than 6 months from the diagnosis. For this reason, early detection is very important, because in low-risk patients treated with auto-HSCT, the 2-year survival in good centers is 94%, and in medium-risk patients treated with classic chemotherapy, it is about 40–50%.22
Transthyretin cardiac amyloidosis is the 2nd most common type of CA. Together, both forms of amyloidosis (AL-CA and ATTR-CA) account for approx. 95% of cases of the disease. Other unique types of amyloidosis are associated with amyloid A and apolipoprotein A.
Transthyretin (TTR) is a physiological protein produced in the liver that transports the thyroid hormone thyroxine (T4) and vitamin A. The essence of ATTR-CA is the instability of the tetramer of which TTR consists.23, 24 The breakdown of the protein molecule causes the formation of misfolded monomers that form insoluble amyloid proteins, which accumulate in the form of deposits in tissues and organs, including the heart.25 The abnormal protein breakdown may be caused by both the mutated TTR gene5 and the spontaneous tetramer breakdown characteristic of wild-type ATTR-CA.25 It has been established in autopsy tests that ATTR-CA may occur in as much as 25% of the elderly population over 85 years of age.5
The accumulation of amyloid in the extracellular space impairs the diastolic function of the myocardium, which is observed as the restrictive cardiomyopathy phenotype. In advanced cases, LV ejection fraction (LVEF) is also impaired. Moreover, amyloid protein deposits disturb the transport of calcium and cell metabolism, and contribute to cardiotoxicity, leading to cellular edema and damage to cardiomyocytes.26
Clinical presentation
Unfortunately, the clinical symptoms of both forms of amyloidosis (AL-CA and ATTR-CA) are not characteristic of this disease, and an early diagnosis is therefore difficult, requires consultations with many specialists and takes a long time,27 often until echocardiography is performed by an experienced cardiologist.28 The most common signs and symptoms of all forms of amyloidosis are listed in Table 1.
Diagnostic procedure
The abnormalities found in CA in non-invasive cardiological diagnostics are presented in Table 2.
Cardiac biomarkers
In everyday cardiological practice, the diagnostic procedure should include the assessment of cardiac markers. Brain natriuretic peptide (BNP), N-terminal prohormone of BNP (NT-proBNP) and troponin T (TnT) are the most widely researched biomarkers of cardiac involvement for the diagnosis of CA.29 The combination of these biomarkers allows also the prognostic stratification. It has been shown that the levels of BNP/NT-proBNP are disproportionately high in CA and are very useful in a prognostic assessment.29, 30 In addition, TnT has a very good prognostic value.31
Electrocardiogram
The analysis of the electrocardiogram (ECG) waveform with a characteristic image of low-voltage QRS complexes and pseudo-myocardial infarction is not a sufficiently sensitive method.32 Low QRS voltage in a patient with echocardiographic myocardial hypertrophy is characteristic of amyloid cardiomyopathy, but is estimated to occur in less than 50% of patients. In addition, atrial fibrillation (AF) and conduction disturbances may also be present.
Echocardiogram
This section refers to Figure 2. The early diagnosis of amyloidosis is greatly facilitated by modern imaging techniques, in particular echocardiography, using the deformation analysis by means of the automatic tracking of myocardial acoustic markers (speckle tracking echocardiography – STE). Global longitudinal strain (GLS) is a non-invasive method to assess the shortening of cardiomyocytes regionally and globally. The technique detects discrete changes in the heart function that cannot be detected with classical echocardiography. In the deformation analysis in patients with amyloidosis, the impaired deformation is observed in the basal and middle segments of the left ventricle as compared to the apical segments. This so-called apical sparing is common in patients with amyloidosis and has a high diagnostic and prognostic value.33, 34 Barros-Gomes et al. showed that GLS most accurately provided additional prognostic information for all-cause mortality as compared to other clinical, echocardiographic and serological indicators.35 Apical sparing is observed both in patients with ATTR-CA and those with AL-CA. The classic manifestations of echocardiographic heart involvement in amyloidosis are shown in Table 2.
Other potentially useful echocardiographic tools for the early detection of abnormalities36 include the LV end-systolic elastance, the LV relaxation time constant – tau, and the assessment of the LV stiffness and the peak left atrial longitudinal strain.37 Moreover, Aimo et al. proposed a simple echocardiographic score to rule out CA.38 The AMYLoidosis Index (AMYLI: relative wall thickness (RWT) × the early mitral inflow velocity (E) to mitral annular early diastolic velocity (e’) ratio (E/e’)) <2.22 excluded heart involvement in that research.38 Other models reported by Boldrini et al. include RWT, the E/e’ ratio, longitudinal strain, and tricuspid annular plane excursion.39 The accuracy of those models was very good, but the results still require external validation.39
Magnetic resonance imaging
of the heart
This section refers to Figure 3. The cardiac morphology and apical sparing specific to echocardiography are also visible in CMR images, the major advantage of which, however, is that they enable tissue characterization of the myocardium. The early deposition of amyloid fibers in the myocardium causes an increase in the extracellular volume (ECV), as can be observed from the presence and location of late gadolinium enhancement (LGE). In the case of amyloidosis, the most common observations are the presence of generalized subendocardial LGE foci, alternatively diffuse subendocardial or even full-thickness lesions.40, 41 There are relatively frequent pseudo-technical difficulties in the so-called complete extinction of the myocardial signal due to the diffuse presence of amyloid. Gadolinium contrast can also be seen in the walls of the enlarged atria and the often thickened atrial septum. New quantitative mapping techniques involve evaluating the native longitudinal relaxation time (T1) and the contrast-enhanced T1, as well as ECV calculated based on them. Similarly to ECV, the native T1 is significantly increased in CA.42
Radioisotope techniques
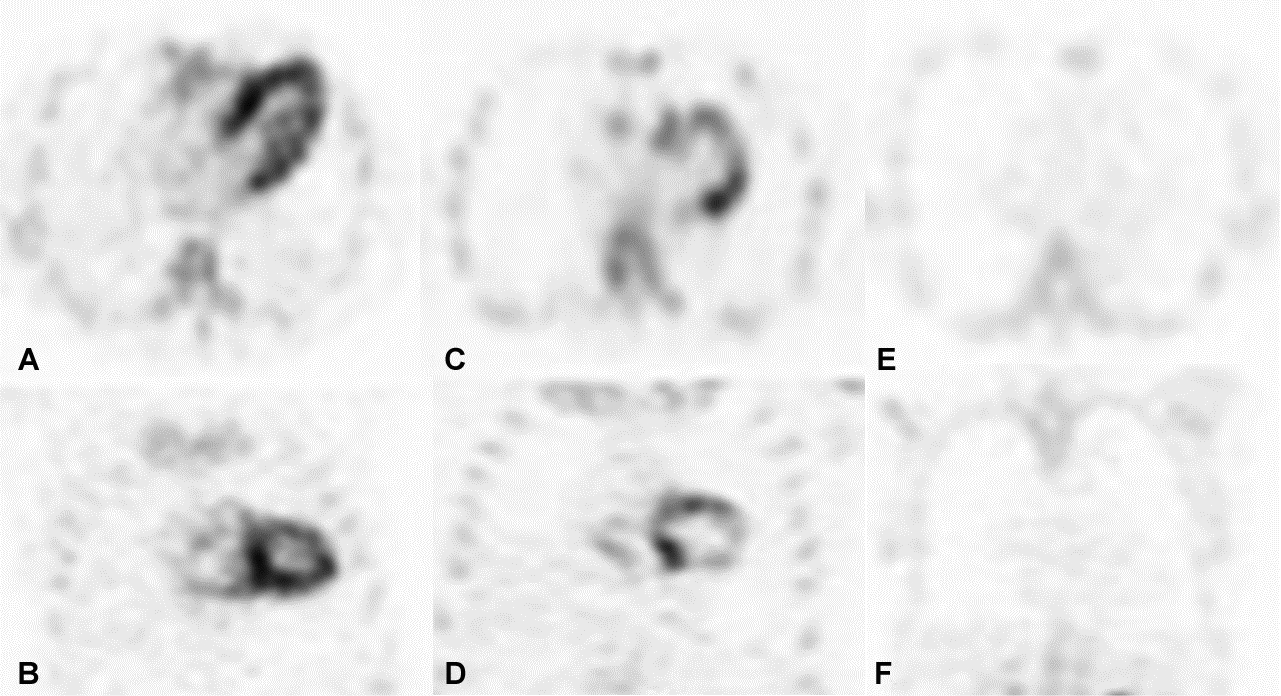
This section refers to Figure 4, Figure 5. Radioisotope techniques are currently the basic method for diagnosing ATTR-CA. The techniques employ the technetium (Tc)-99m radioisotope and the tracers classically used in the skeletal examination: 3,3-diphosphono-1,2 propanodicarboxylic acid (DPD); pyrophosphate (PYP); and methylenediphosphonic acid (MDP).43 However, scintigraphy is often available only in reference centers. The simplicity of imaging and a high specificity of nearly 100% (in the absence of light chains and monoclonal protein) for ATTR-CA are the advantages of scintigraphy emphasized by many authors.44, 45, 46 Currently, a semi-quantitative visual assessment is applied using the Perugini 4-point scale (0–3).44 The degree of isotope accumulation is compared to the flat sections of the ribs. What is characteristic of ATTR-CA is the diffuse uptake of the radioisotope in the myocardium. A complete scintigraphy analysis of the heart may also include the interpretation of the single-photon emission computed tomography (SPECT) perfusion as well as the calculation of the heart/contralateral lung (H/CL) index. This index is a quantitative comparison of the region of interest (ROI) ratio in the heart and the contralateral lung tested after 1 h (>1.5) or 3 h (>1.3).47 The observations of Gillmore et al. confirmed the considerable usefulness of scintigraphy with the abovementioned bone tracers.48 In the absence of monoclonal protein in serum or urine, and with the grade 2 or 3 uptake of the radioisotope, the specificity of the ATTR-CA diagnosis with this method was 100% in that publication. The authors concluded that it was unnecessary to perform an endomyocardial biopsy.48 Furthermore, Marume et al. assessed a combination of parameters to increase the pretest probability of scintigraphy.49
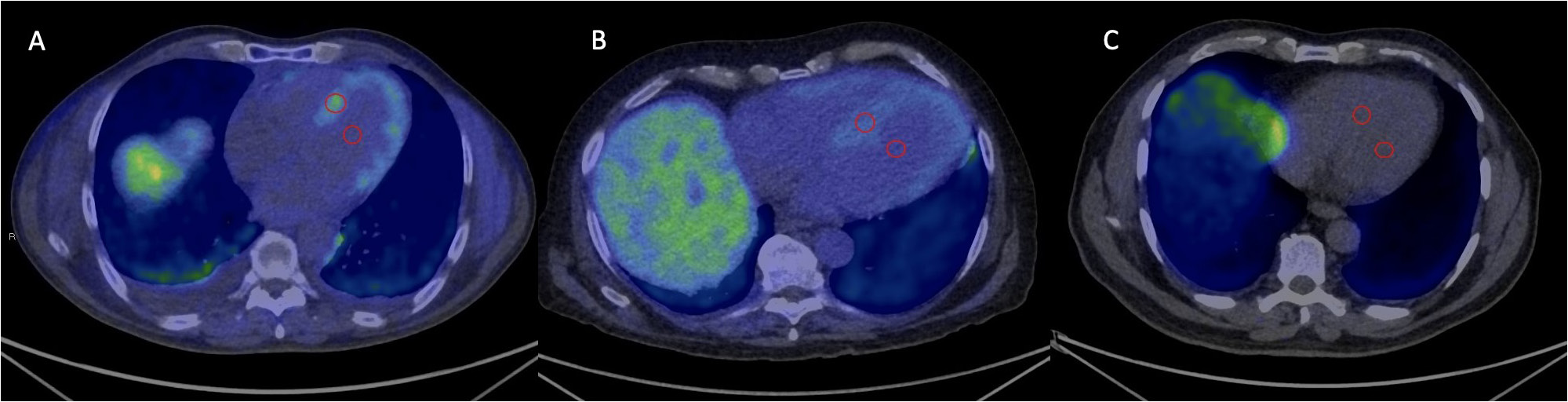
Positron emission tomography (PET) using the Pittsburgh compound-B (PiB) radiotracer is another recommended method of examination that uses radiotracers. Pittsburgh compound-B is a radioactive 11C-labeled analog of thioflavin T, originally used for imaging beta-amyloid plaques in the brain tissue in the diagnosis of Alzheimer’s disease. The PiB radiotracer binds directly to the amyloid protein in both ATTR-CA and AL-CA. The myocardial uptake is quantified in relation to the blood activity of the heart cavities, adopting different breakpoints for the 2 types of amyloidosis. The normal myocardial/blood uptake ratio is approx. 1.50
Machine learning models
The application of artificial intelligence for screening CA has been reported recently.51, 52 The authors showed that machine learning models well identified patients with CA.
Therapeutic procedure
There are 3 distinct areas of treatment for amyloidosis.
The 1st area of treatment is the management of cardiac complications due to the accumulation of abnormal protein throughout the heart, causing the symptoms of HF and impairing the exercise capacity, but also potentially leading to atrioventricular conduction disorders or supraventricular tachyarrhythmias.
Angiotensin receptor antagonists in low doses may be profitable, as they act as afterload-reducing agents, and improve the forward cardiac flow and renal perfusion. Similarly, low doses of β-blockers are helpful as rate-controlling agents. However, the use of medicines typical for HF, such as angiotensin-converting enzyme inhibitors (ACEIs), β-blockers or angiotensin receptor–neprilysin inhibitors (ARNIs), is not recommended in advanced heart involvement. On this level, the adverse effects of these agents (hypotension, kidney failure, conduction disorders) overwhelm benefits.52 The use of calcium antagonists, particularly non-dihydropyridine calcium channel blocker and digoxin is also avoided.53
The 2nd area of treatment is reserved for hematologists and concerns AL-CA. Depending on the patient’s classification to the appropriate risk group, the most commonly used regimens are immunochemotherapy based on bortezomib, cyclophosphamide and melphalan. Selected patients undergo high-dose therapy supported with auto-HSCT.54
The 3rd area of therapy concerns patients with ATTR-CA. The treatment of this specific pathology involves 3 management strategies. These are as follows: the inhibition of protein production in the liver; the stabilization of the tetramer; and the disruption of the permanent bonding of amyloid fibers.
The 1st strategy of ATTR-CA management, which involves impairing the production of abnormal protein in the liver, associated with the genetically determined form of ATTR-CA, can now be performed with the use of gene therapy approved by the U.S. Food and Drug Administration (FDA), applying an RNA interference therapeutic agent – patisiran55 or an antisense oligonucleotide – inotersen.56 Both drugs are registered for amyloid polyneuropathy, but not for ATTR-CA. The estimated annual cost of such therapy in the USA is currently around $300,000. Liver transplantation is also performed less and less often for these indications.
The stabilization of the TTR tetramer is possible with the drug tafamidis, which binds selectively to the 4-part TTR oligomer. The Transthyretin Amyloidosis Cardiomyopathy Clinical Trial (ATTR-ACT) showed a significant decrease in the number of hospitalizations and mortality of patients treated with tafamidis.57 In 2019, the drug was also approved in the USA for ATTR-CA. Another TTR tetramer stabilizer, diflunisal, is a non-steroidal anti-inflammatory drug, but its use was not associated with an improved prognosis in patients with cardiac involvement in randomized studies and was accompanied by numerous complications, primarily from the gastrointestinal tract.58, 59, 60
Another therapeutic strategy in ATTR-CA is an attempt to amyloid degradation with doxycycline or tauroursodeoxycholic acid. However, the effects of this procedure require a further assessment. There are also clinical trials with the use of a specific monoclonal antibody PRX 004 affecting amyloid degradation.61 In extreme cases, heart and liver transplants are performed.
Conclusions
ATTR-CA ‘timeline’
An interesting timeline for the progression of ATTR-CA was proposed by the authors from the Mayo Clinic.62 The presence of bilateral carpal tunnel syndrome, the stenosis of the lumbar spine or the rupture of the biceps tendon, which may appear many years before the disease manifestation, are considered to be extremely early clinical symptoms (“red flags”) of the disease. After some time, there may be abnormalities in PET/CT, SPECT/CT and CMR imaging tests with normal cardiac biomarkers and no morphological changes in echocardiography. On the other hand, typical changes in the echocardiographic examination and other clinical symptoms indicate that the disease is highly advanced and they clearly manifest the full-blown disease. It has been noticed that ECG abnormalities become apparent only at a very late stage.
Algorithm for assessing a patient
with suspected amyloidosis
The authors of this study use a modified algorithm for assessing a patient with suspected amyloidosis (Figure 6).3, 48, 62 It primarily includes the echocardiographic or CMR assessment using GLS as well as the laboratory assessment of concentrations and the ratio of free light chains (FLCs) and immunofixation, in both serum and urine, to exclude AL-CA.
If there is 1 or more of these 3 protein laboratory abnormalities, then the patient should be referred for further hematological evaluation. A typical picture of bone marrow biopsy changes or Congo red-stained amyloid confirm the diagnosis of AL-CA with high probability. If scintigraphy shows in the patient the cardiac uptake (grade 1–3), monoclonal gammopathy of undetermined significance (MGUS) or AL-CA co-occurrence with ATTR-CA should be considered.3
If the abovementioned proteins are not detected, the SPECT/CT examination is performed with the use of DPD or PYP labeled with technetium-99m (Tc-99m). Grade 2 or 3 uptake in the absence of light chains and monoclonal protein allows the diagnosis of ATTR-CA. In doubtful cases (scintigraphy grade 0–1), when both AL-CA and ATTR-CA are highly suspected, patients should be additionally referred for CMR or PET/CT. A myocardial biopsy and the analysis of amyloid protein composition using mass spectrometry should also be considered.