Abstract
The coexistence of mosaic trisomy 18 and Turner syndrome represents an extremely rare genetic constellation, with only a few cases reported in the literature to date. We present the case of a 7-year-old girl with esophageal atresia, full trisomy 18 in lymphocytes, and mosaic copy number changes involving trisomy 18 with coexisting monosomy X in fibroblasts, suggesting the presence of additional cell populations lacking trisomy 18. In addition, we provide a review of the relevant literature. This case is of particular clinical significance, as it describes long-term survival in a child with Edwards syndrome and illustrates the diagnostic challenges associated with tissue-limited mosaicism. Furthermore, it underscores that an established diagnosis of one genetic disorder does not preclude the coexistence of another, particularly when the clinical phenotype deviates from the classical presentation of the primary syndrome. Adequate analysis of a sufficient number of metaphases in karyotyping is essential to reliably detect or exclude mosaicism in Edwards syndrome, as the distinction between full and mosaic trisomy 18 carries important prognostic implications.
Key words: trisomy 18 syndrome, Turner syndrome, mosaicism, esophageal atresia, chromosomal aberrations
Introduction
Mosaicism is a phenomenon in which distinct cell lineages carrying different genomes coexist within a single organism. Germline mosaicism is restricted to germ cells and is associated with heritability, whereas somatic mosaicism involves the remaining somatic cells and may contribute to the manifestation of a specific phenotype in an individual patient. Depending on the developmental stage at which a somatic mutation arises, the affected cells may be distributed throughout the entire organism or confined to particular tissues or organs, which can directly influence the severity of symptoms associated with the given genotype. In addition to situations in which a cell line carrying a chromosomal aberration coexists with a normal cell line, double mosaicism may also occur, in which both cell lines exhibit different types of chromosomal aberrations. This may result in the overlap of 2 distinct abnormal phenotypes, significantly complicating the patient’s clinical presentation.1
Edwards syndrome is a severe chromosomal disorder caused by the presence of an extra chromosome 18. The condition occurs in approx. 1 in 6,000 live births. Of all trisomy 18 cases, 94% represent full trisomy (simple or translocation-associated), while 5% involve mosaicism, in which both normal and trisomy 18 cell lines are present. Suspicion of Edwards syndrome is typically raised prenatally or during the neonatal period based on characteristic dysmorphic features and prenatal-onset growth restriction, whereas survival is determined mainly by the severity of organ malformations, particularly congenital heart defects, as well as respiratory and feeding difficulties. The most common congenital malformations involve the cardiovascular and genitourinary systems. Gastrointestinal anomalies, such as esophageal atresia with tracheoesophageal fistula, are not a typical feature of trisomy 18 but occur with increased frequency compared with the general population. Severe psychomotor and cognitive developmental delay is typically present. However, the clinical presentation of mosaic trisomy 18 is highly variable, ranging from the complete trisomy 18 phenotype to the absence of dysmorphic features and preserved cognitive function. Approximately 50% of infants with trisomy 18 survive longer than 1 week, and 5–10% survive beyond the 1st year of life. Mortality is primarily associated with congenital heart defects, apnea, and recurrent respiratory infections. Survival is also strongly influenced by gestational age at delivery.2, 3
Turner syndrome is a genetic disorder caused by the complete or partial loss of one X chromosome, leading to characteristic phenotypic features such as short stature, gonadal dysgenesis, and cardiovascular and renal abnormalities. The condition occurs in approx. 1 in 2,500 live-born females. Among all Turner syndrome cases, approx. 40–50% involve monosomy X (45,X), while mosaic forms account for about 28–40%, and structural X chromosome abnormalities represent approx. 15% of cases.4 The simultaneous occurrence of both Turner syndrome and Edwards syndrome in mosaic form is an extremely rare phenomenon, with only a handful of cases documented in the medical literature. These rare reports suggest that the clinical presentation of such mosaic cases is highly variable, depending on the proportion of different cell lines and the extent of tissue involvement.
Below, we present the case of a 7-year-old girl with esophageal atresia, full trisomy 18 in lymphocytes, and mosaic copy number changes involving trisomy 18 with coexisting monosomy X in fibroblasts, suggesting the presence of additional cell populations lacking trisomy 18. This case is of particular clinical significance, as it describes a child with Edwards syndrome who demonstrated long-term survival and in whom an initial finding of trisomy 18 was later clarified through analysis of a 2nd tissue sample, revealing mosaicism.
A literature search was performed in PubMed using combinations of the terms “45,X” and “47,XX,+18”. A total of 8 relevant case reports describing mosaicism involving both 45,X and 47,XX,+18 cell lines were identified and included in the analysis.5, 6, 7, 8, 9, 10, 11, 12
Ethical considerations
Written informed consent was obtained from the patient’s parents for publication of this paper. The patient’s parents also provided informed consent for genetic testing, publication of this work, and presentation of their child’s image. The study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the Ethics Committee of Wroclaw Medical University, Wrocław, Poland (approval No. KB 430/2018).
Clinical presentation
The patient was born as the 3rd child to non-consanguineous parents (mother aged 36 years, father aged 37 years) following an uncomplicated vaginal delivery at 39 weeks of gestation. Birth weight was 2,390 g (standard deviation score (SDS) = −3.03 for gestational age), length 51 cm (SDS = 0.67 for gestational age), and head circumference 30 cm (SDS = −3.3), consistent with microcephaly. The Apgar score was 10. The pregnancy was complicated by polyhydramnios, a cardiac septal defect, and intrauterine growth restriction detected on prenatal ultrasonography.
On the 1st day of life, the girl underwent surgery for esophageal atresia with tracheoesophageal fistula. Postoperatively, she required intensive care due to sepsis and cardiorespiratory failure. Because of the cardiac septal defect, esophageal atresia, growth restriction, and dysmorphic features, cytogenetic analysis of peripheral blood lymphocytes was performed. Karyotype analysis, based on the evaluation of 15 metaphases, revealed 47,XX,+18 (the abnormality was present in cells from 2 independently established peripheral blood lymphocyte cultures). Following the genetic diagnosis, a care plan addressing the potential futility of further life-sustaining treatment was developed. The patient was extubated on day 10 and remained respiratory stable thereafter. In the following years, psychomotor developmental delay was observed, with sitting, walking, and speech milestones achieved at 10, 23, and 36 months of age, respectively. Between the 2nd and 3rd years of life, the patient experienced 3 episodes of lower gastrointestinal bleeding, which required hospitalization on 2 occasions. Infectious causes were clinically excluded. Technetium-99m pertechnetate scintigraphy was performed due to suspected Meckel’s diverticulum, but the study revealed no focal tracer uptake suggestive of ectopic gastric mucosa. Additionally, abdominal ultrasonography revealed a focal hepatic lesion, which has remained under observation. The patient remained under regular follow-up in multiple specialist clinics. Cardiology follow-up revealed a hemodynamically insignificant ventricular septal defect; the atrial septal defect present at birth closed spontaneously. Orthopedic assessment identified right hip dysplasia and scoliosis associated with a congenital L3 hemivertebra. Ophthalmologic evaluation confirmed strabismus. Otolaryngologic follow-up documented a history of otitis media associated with hearing impairment. Renal ultrasonography findings were normal. At the age of 4 years, she underwent surgical excision of an epidermal cyst located in the suprasternal region. Due to the patient’s prolonged survival, genetic testing was repeated using fibroblasts obtained during the surgical procedure. Array comparative genomic hybridization (array-CGH) revealed mosaic copy number changes involving trisomy 18 and monosomy X: arr(X)x1[0.35],(18)x3[0.65], suggesting the possible presence of a cell population with a normal copy number of chromosome 18.
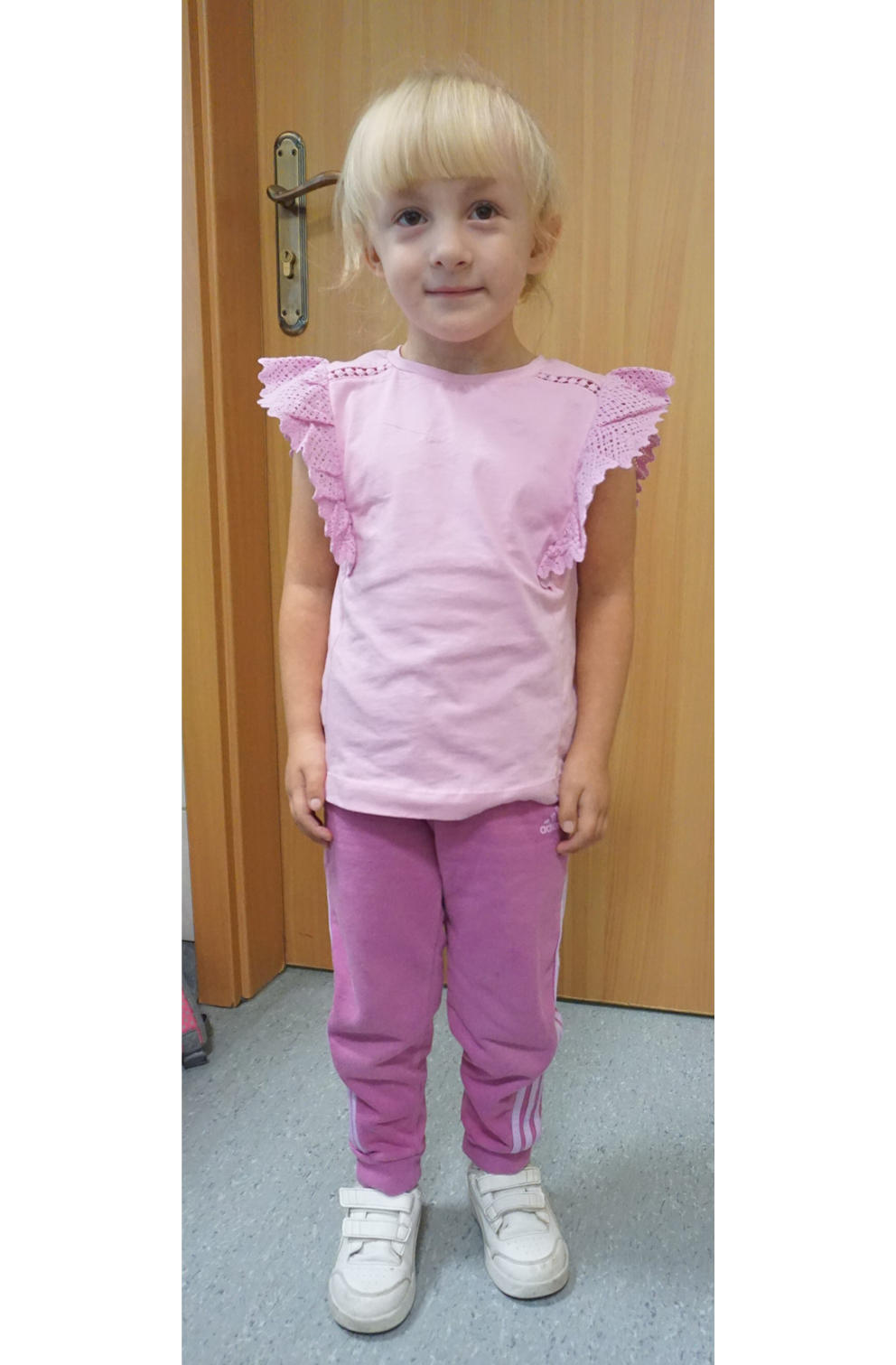
At the age of 6.5 years, the girl was referred to the Department of Pediatric Endocrinology with a diagnosis of mosaic Edwards syndrome and mosaic Turner syndrome. From early childhood, growth retardation and breast enlargement had been observed. Ultrasonography performed at 2 years of age confirmed the presence of glandular breast tissue. The patient was diagnosed with mild intellectual disability and is currently enrolled in an early developmental support program. On admission, physical examination revealed short stature, gait with adduction of the right forefoot, scoliosis, asymmetry of the palpebral fissures (with the right fissure being narrower), and strabismus. Pubertal status was assessed as Tanner stage B3/P1. Auxological examination revealed a height of 105 cm (<3rd percentile; height SDS = −2.85), weight of 19.2 kg, and body mass index (BMI) of 17.4 kg/m2 (75–90th percentile; SDS = 0.5). The patient’s general appearance is shown in Figure 1. Bone age was assessed as 6 years at a chronological age of 6.5 years. Hormonal parameters, including estradiol, follicle-stimulating hormone (FSH), and luteinizing hormone (LH), were within normal limits (estradiol <20.0 pg/mL; FSH 2.01 mIU/mL, reference range 0.5–3.2; LH <0.1 mIU/mL, reference range <0.3 mIU/mL). Pelvic ultrasonography revealed a uterine structure with dimensions at the upper limit of normal for age. The ovaries were not visualized.
Discussion
One of the most striking features in our patient is the relatively favorable clinical course and long-term survival, which are highly atypical for classic trisomy 18, in which survival beyond the 1st year is rare. An earlier study published in 1994 suggested that prolonged survival in children with full trisomy 18 may reflect natural variability in disease severity rather than undetected mosaicism. However, more recent studies consistently demonstrate that mosaic trisomy 18 is associated with a milder clinical presentation and improved survival. For example, an analysis based on parent questionnaires and medical records from 98 families showed that children with mosaic trisomy 18 exhibit a distinctly milder phenotype compared with those with full trisomy 18.13 A subsequent study analyzing 326 live births with trisomy 18 (309 full trisomy and 17 mosaic cases) demonstrated that early survival is substantially higher in mosaic cases: approx. 70% survive the 1st year, and 41% survive at least 5 years, compared with 8% and 1%, respectively, in full trisomy 18.14
Another study published in 2025 reported that, in a cohort of 503 patients, 8.5% survived to the age of 10 years. After distinguishing between mosaic/partial trisomy 18 (considered jointly) and full trisomy 18, approx. 44% of individuals in the former group reached 10 years of age, whereas only 6.7% in the latter group did. It remains unclear whether these results were based solely on peripheral blood lymphocyte analysis or whether mosaicism may have been absent in blood but present in other tissues.15
Based on the review of the 8 analyzed articles, it was found that in 2 cases, the initial karyotype analysis of peripheral blood lymphocytes (evaluating 16 and 50 metaphases, respectively) suggested full trisomy 18. In both cases, however, the clinical presentation was atypical for Edwards syndrome, prompting further diagnostic evaluation. Subsequent extended investigations, including fibroblast analysis and fluorescence in situ hybridization (FISH), revealed mosaicism with a lower proportion of trisomy 18 cells; in 1 case, an additional ring chromosome 18 was identified.10, 12 In our case, the situation was somewhat different. The patient presented with phenotypic features consistent with Edwards syndrome; however, prolonged survival and a relatively milder clinical course were atypical for full trisomy 18.
Mosaic trisomy 18 is associated with longer survival and a milder phenotype; therefore, analysis of a sufficient number of metaphases to detect or exclude mosaicism is essential, as distinguishing mosaic from full trisomy 18 carries important prognostic significance. Our patient was initially diagnosed with full trisomy 18 based on karyotype analysis of peripheral blood lymphocytes. Mosaicism was subsequently demonstrated by array-GCH analysis of skin fibroblasts. The estimated 65% trisomic signal indicates that approx. 35% of the cell population does not harbor trisomy 18. In addition, the array profile suggests that approx. 35% of the cell population lacks 1 X chromosome.
The diagnosis of 1 genetic disorder does not exclude the presence of another, and this possibility should be considered, especially when the patient’s clinical phenotype deviates from the typical spectrum of the primary syndrome.16, 17 According to current diagnostic guidelines, the diagnosis of Turner syndrome requires the presence of X chromosome abnormalities together with characteristic phenotypic features.4
The coexistence of mosaic Turner syndrome and trisomy 18 in a single patient represents an extremely rare genetic constellation, with very few cases documented in the literature to date.5, 6, 7, 8, 9, 10, 11, 12 The phenotype of these patients is highly variable, ranging from severe presentations with predominant features of trisomy 18 to relatively mild manifestations, and depends on the proportions and tissue distribution of the different cell lineages. However, the distribution of trisomic cells in critical organs cannot be predicted based on peripheral blood findings.18
The clinical presentation is not simply a composite of trisomy 18 and Turner syndrome but rather reflects the dominant karyotypic lineage in key tissues. In a case reported by Schluth-Bolard et al., cytogenetic analysis was performed for reasons unrelated to clinical suspicion of trisomy 18 or Turner syndrome, and the diagnosis was established only after genetic testing, followed by retrospective phenotypic evaluation. Cytogenetic and FISH studies revealed 45,X/47,XX,+18 mosaicism with clear tissue dependence, characterized by a predominance of monosomy X in skin fibroblasts and trisomy 18 in peripheral blood cells. This finding represented a rare example of tissue-specific mosaicism associated with distinctive facial features, bicuspid aortic valve, and ovarian agenesis.10 Previous reports indicate that in 45,X/47,XX,+18 mosaicism, trisomy 18 tends to predominate in peripheral blood, whereas monosomy X is more prevalent in other tissues, consistent with the genetic findings observed in our patient.
This exceptional rarity presents significant diagnostic and therapeutic challenges. From the perspective of phenotype–genotype correlation, dual mosaicism may sometimes help explain the mixed clinical picture: several findings align with trisomy 18, whereas others may reflect the influence of Turner syndrome. Difficulties arise when a particular feature (e.g., short stature in our case) is observed in both syndromes. Impaired growth in Turner syndrome is caused by haploinsufficiency of the SHOX (short stature homeobox) gene located on the X chromosome and requires treatment with recombinant growth hormone at supraphysiological doses. Growth retardation in Edwards syndrome is common and multifactorial, resulting from intrauterine growth restriction, severe congenital anomalies, feeding difficulties, and chronic cardiorespiratory failure. Its exact pathogenesis has not been fully elucidated. From a clinical perspective, management of Edwards syndrome is primarily determined by the severity of congenital defects and the high mortality in early life rather than by growth impairment itself. In the presence of full trisomy 18 in peripheral blood and mosaic copy number changes involving trisomy 18 with coexisting monosomy X in fibroblasts, with a phenotype clearly dominated by Edwards syndrome rather than Turner-related features, and in the context of concurrent scoliosis, there are no justified indications for initiating growth hormone therapy.
Cardiac defects suggestive of Turner syndrome typically involve left-sided cardiac structures, whereas a ventricular septal defect (VSD) is less characteristic. A VSD may be considered part of the Turner syndrome phenotype if accompanied by other typical features,4 but it is more commonly associated with Edwards syndrome. Our patient presented with thelarche praecox, an atypical finding in Turner syndrome, in which spontaneous breast development is usually absent during puberty, whereas in mosaic Turner syndrome it may occur at the expected pubertal age. Isolated premature thelarche is defined as the development of breast tissue in the absence of other signs of pubertal development, such as accelerated growth velocity, rapid breast progression, or advanced skeletal maturation.19 In pediatric patients, non-visualization of the ovaries on ultrasonography may reflect technical limitations or small gonadal size rather than true ovarian agenesis. Accordingly, thelarche in our patient may be consistent with isolated premature thelarche, a condition that can occur in otherwise healthy girls; in this case, it may be related to mosaic gonadal cell lines with preserved ovarian function.
In patients with mosaicism, genetic testing limited to a single cell type, such as peripheral blood lymphocytes, may be insufficient for detecting a pathogenic variant. The distribution and proportion of mutant cells across different tissues depend on several biological and technical factors.
First, the embryological origin of different tissues plays a crucial role. During early development, cells differentiate into distinct germ layers (ectoderm, mesoderm, and endoderm) which give rise to separate tissue lineages. Since blood cells originate primarily from the mesoderm, whereas tissues such as skin or oral epithelium derive from other germ layers, a postzygotic mutation may be confined to a single lineage, resulting in lineage-specific mosaicism detectable only in certain tissues.20
Second, cellular selection may influence the detectability of mosaicism. Cells carrying chromosomal or genomic aberrations may have reduced viability or proliferative potential in particular tissues, such as peripheral blood, leading to their gradual elimination. Consequently, the abnormal clone may persist in other tissues (e.g., gonadal or cutaneous cells) while being absent from blood-derived DNA, resulting in a false-negative lymphocyte-based result.
Third, technical limitations of genetic testing may contribute to underdetection. Conventional karyotype analysis typically examines a limited number of metaphase cells, resulting in relatively low sensitivity for detecting low-level mosaicism (<10%).
Finally, the timing of the postzygotic mutation affects the extent of its distribution. Mutations occurring later in embryogenesis tend to affect a smaller proportion of tissues, potentially sparing hematopoietic cells altogether.21, 22
For these reasons, when the clinical presentation strongly suggests a genetic disorder but testing of a single cell type (e.g., lymphocytes) yields negative or inconclusive results, it is advisable to repeat the analysis using DNA from an alternative tissue source, such as fibroblasts, buccal epithelial cells, or urinary epithelial cells. This strategy increases the likelihood of detecting tissue-specific or low-level mosaic abnormalities and facilitates a more accurate molecular diagnosis.
Limitations of the study
This report is limited by evaluating mosaicism solely in peripheral tissues and lacking a classical fibroblast karyotype. Although expanding the phenotypic spectrum of trisomy 18 mosaicism, clinical findings must guide prognosis rather than peripheral cytogenetic percentages.
Conclusions
Analysis of a sufficient number of metaphases is essential to reliably detect or exclude mosaicism in Edwards syndrome, as the distinction between full and mosaic trisomy 18 carries important prognostic implications. When the clinical presentation is atypical, repeat analysis using biological material derived from an alternative tissue source should be considered. The diagnosis of one genetic disorder does not exclude the presence of another, and this possibility should be considered, particularly when the patient’s clinical phenotype deviates from the typical spectrum of the primary syndrome.
Use of AI and AI-assisted technologies
ChatGPT 5 were used for language editing, stylistic refinement, and improvement of clarity.