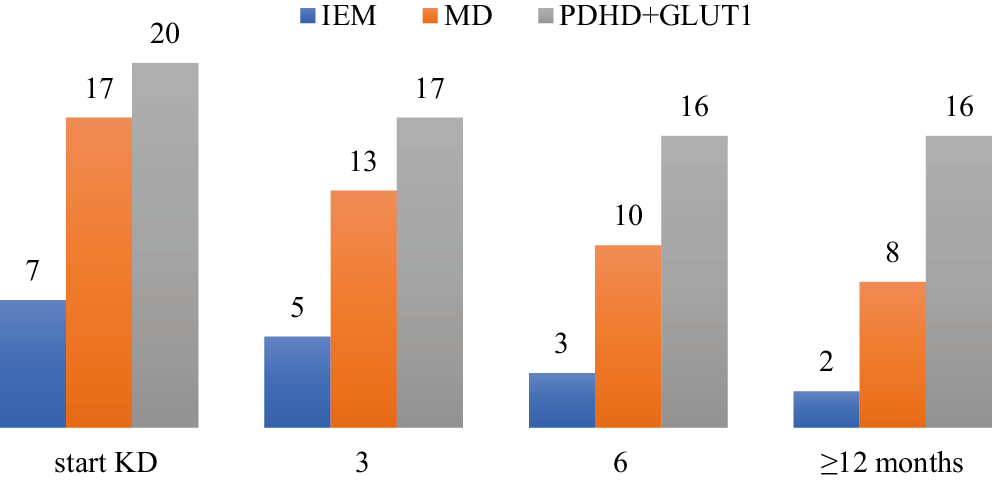
Abstract
Background. The ketogenic diet (KD) is an established therapeutic option for epilepsy and selected inborn errors of metabolism (IEMs), particularly glucose transporter type 1 deficiency (GLUT1D) and pyruvate dehydrogenase complex deficiency (PDCD). Increasing evidence suggests broader applications of KD in pediatric metabolic disorders; however, data on its safety and efficacy in heterogeneous IEM populations remain limited.
Objectives. To evaluate the efficacy, clinical benefits, and adverse effects (AEs) of KD in pediatric patients with various IEMs.
Materials and methods. A retrospective analysis was conducted in pediatric patients with IEMs receiving KD treatment. Patients were categorized into 3 groups: 1) other IEMs (n = 7), 2) mitochondrial diseases (MD) (n = 17), and 3) GLUT1D and PDCD (n = 20). The median age at initiation of KD was 37, 53, and 53 months, respectively, and the median duration of KD treatment was 5, 11, and 55 months in groups 1, 2, and 3.
Results. The KD was associated with clinical benefits in 84% of patients. Among children with epilepsy (n = 23), a seizure reduction of >50% was observed in 73.9% of patients, including complete seizure freedom in 4 individuals. Improvements were also noted in muscle tone (27.6%), exercise tolerance (51.2%), ataxia (83.3%), and involuntary movements (60%). Lactate levels decreased in 84.6% of patients with mitochondrial disease and in all patients with PDCD. The KD was discontinued in 12 patients due to insufficient efficacy (n = 5) or AEs (AEs; n = 7). The most common AEs included gastrointestinal (GI) symptoms, dyslipidemia, hyperuricemia, metabolic acidosis, and decreased free carnitine; most were transient. No significant association was found between median β-hydroxybutyrate (BHB) levels and clinical outcomes.
Conclusions. The KD is an effective and generally well-tolerated therapeutic option in pediatric IEMs, with benefits extending beyond seizure control. Adverse effects are typically manageable, although GI intolerance may limit long-term use. Ketogenic diet should be considered not only for refractory epilepsy but also for selected metabolic indications.
Key words: ketogenic diet, inborn errors of metabolism, pediatric metabolic disorders, mitochondrial diseases, epilepsy
Background
The ketogenic diet (KD) is a high-fat, low-carbohydrate dietary regimen that induces ketone body production by restricting carbohydrate intake in favor of fat. Several KD variants are used in clinical practice: the classical KD, modified Atkins diet (MAD), medium-chain triglyceride (MCT) diet, and low-glycemic index treatment.1 Although originally introduced for epilepsy, the KD is now being considered in many neurological and metabolic diseases.
The mechanisms of the KD have not been fully elucidated, but a number of potentially beneficial effects have been identified. Ketone bodies, produced in excess during the KD, serve as an alternative energy substrate for cells, including those of the central nervous system (CNS). They modify neurotransmission, reduce neuronal membrane excitability, and influence dopamine synthesis and signaling. The ketogenic diet also modulates cellular energy pathways, reduces oxidative stress and free radical production, exerts anti-inflammatory effects, promotes remyelination, and is believed to reduce epileptogenesis.2, 3 Ketone bodies likely stimulate mitochondrial biogenesis, influence mitochondrial dynamics and elongation, and activate mitochondrial autophagy, contributing to the removal of dysfunctional mitochondria. In addition, some MCTs used in the KD interact with receptors in the CNS, including inhibition of AMPA glutamate receptors, and are particularly effective in promoting ketogenesis.4
The KD has a well-established role in epilepsy, especially in drug-resistant cases.1, 5, 6 Beyond epilepsy, growing evidence supports the KD in several neurological disorders, such as neurodegenerative diseases, multiple sclerosis, migraine, traumatic brain injury, and chronic pain.7, 8, 9 It has also been investigated in non-neurological conditions, including depression, polycystic ovary syndrome, diabetes, and CNS tumors.3, 10, 11
Pyruvate dehydrogenase complex deficiency (PDCD) and glucose transporter type 1 deficiency (GLUT1D) are inborn errors of metabolism (IEMs) in which the KD is considered the treatment of choice and is usually recommended as long-term or lifelong therapy. Its effectiveness in these conditions has been demonstrated in numerous reports.12, 13, 14 By contrast, the use of the KD in other IEMs is reported only sporadically, mostly as single case reports or small series.2, 15 Among more than 1,500 described IEMs, approx. 1/3 are associated with epileptic seizures, which are often resistant to standard pharmacological treatment. As a result, metabolic epileptic encephalopathies arising from IEMs could be treated with the KD.1
When the KD is used in IEMs, different mechanisms of action can be exploited. In some disorders, such as GLUT1D or mitochondrial diseases (MD), ketones serve as an alternative fuel for the CNS or other tissues. In non-ketotic hyperglycinemia, reducing dietary glycine intake may be beneficial, whereas in glycogen storage diseases, lowering carbohydrate load and providing fat as an alternative energy source play a central role.1
Across various indications, numerous mainly retrospective studies and case reports/series suggest that the KD is generally safe and well tolerated.14 The most common adverse effects (AEs) include metabolic acidosis, hypoglycemia, gastrointestinal (GI) complaints, hyperlipidemia, and an increased risk of kidney stones.1
Objectives
The primary aim of this study was to retrospectively analyze the efficacy and clinical benefits of the KD, as well as the incidence and nature of AEs, in pediatric patients (<18 years) with IEMs treated with the KD.
Material and methods
Study population and grouping
Children with IEMs who were hospitalized and treated at the Department of Pediatrics, Nutrition, and Metabolic Diseases (Children’s Memorial Health Institute, Warsaw, Poland) between January 2015 and June 2025 were included in the study. Patients were divided into 3 groups: group 1 – other IEMs (various metabolic disorders not classified as MD, GLUT1D, or PDCD); group 2 – MD; group 3 – GLUT1D and PDCD. All patients, except 1 child with MD, had a molecularly confirmed diagnosis (Supplementary Table 1).
Data collection and clinical parameters
The following were analyzed: age at KD initiation, duration of treatment, clinical and biochemical benefits of the KD, and the occurrence of AEs, primarily metabolic acidosis (defined as venous blood pH < 7.35 and SE <−5), hypertriglyceridemia (triglycerides >150 mg/dL), hypercholesterolemia (total cholesterol >190 mg/dL), hyperuricemia (uric acid >5 mg/dL), changes in abdominal ultrasound, electrocardiographic (ECG) abnormalities, and GI disorders. All available laboratory results from the medical records were included. For each parameter, the maximum abnormal value over the entire KD treatment period was recorded, and whether the abnormality resolved or persisted was noted. Laboratory testing (including glucose, blood gases, lipids, liver enzymes, urea, and creatinine) was performed at every follow-up visit (every 3–6 months). Electrocardiography and abdominal ultrasound were performed every 6–12 months, or more often if clinically indicated.
Ketogenic diet initiation and dietary management
All patients were under both medical and dietary supervision. The KD was initiated during hospitalization after clear indications were established and contraindications excluded (e.g., fatty acid oxidation defects, disorders of carnitine metabolism, porphyria). On the first day, patients received a 1:1 diet (for every gram of fat, 1 g of protein plus carbohydrates in total). Caloric intake was calculated based on ideal body weight norms for age and sex. Protein intake was within normal ranges, and fluid intake was not restricted. Fasting was not used at diet initiation.
Over the subsequent days, the fat ratio was gradually increased to 1.5:1 and then to 2:1, aiming to achieve therapeutic ketosis (up to a maximum ratio of 4:1). A therapeutic level of ketone bodies was defined as a serum β-hydroxybutyrate (BHB) concentration of 2–5 mmol/L. For patients on the MAD, carbohydrate intake was restricted to 20 g/day; in these cases, serum BHB often remained below 2 mmol/L. During hospital initiation, biochemical parameters and clinical tolerance of meals were closely monitored. Individual menus were developed taking into account dietary preferences, age, and body weight.
Follow-up and ketosis monitoring
The first follow-up visit after KD initiation was usually scheduled 4–6 weeks later. Subsequent visits occurred every 3 months during the first 2 years and every 3–6 months thereafter. At each visit, a clinical dietitian reviewed a 3-day dietary record, recalculated the KD ratio, assessed adherence to dietary recommendations, and verified the correctness and completeness of the diet. In cases of insufficient ketosis (BHB < 2 mmol/L, despite therapeutic targets), the diet was modified – e.g., by adjusting the fat ratio or adding MCT oil to meals at 1 mL/kg/day. For each patient, the median BHB concentration was calculated using all available serum BHB values measured at the site during scheduled visits. Home BHB measurements were not included in the analysis.
Indications and treatment discontinuation
Indications for the KD were: 1) confirmed GLUT1D or PDCD (the KD as standard treatment); 2) epilepsy associated with other IEMs; 3) glycogen storage disease type IIIa (GSDIIIa); 4) MD, with or without seizures. Effectiveness was assessed based on parental reports and clinical evaluation by the treating physicians. No standardized scales were used to quantify muscle tone, exercise tolerance, involuntary movements, or functional independence. The diet was discontinued if, after a minimum of 3 months of therapy, the treatment was ineffective, if serious AEs occurred, if the patient did not tolerate the therapy well, or in the event of the patient’s death.
Statistical analyses
All statistical analyses were performed using GraphPad Prism v. 10.6.1 (GraphPad Software, San Diego, USA). The variables presented and analyzed were few, so their distributions were assumed to be non-normal, and nonparametric tests were used. In case of doubt regarding the distribution of characteristics (e.g., treatment duration across all patients), the Shapiro–Wilk test was used to assess normality (none of the characteristics presented in this study were normally distributed). Time-related differences were assessed using a rank-based repeated-measures test (Kruskal–Wallis). Correlations between variables (e.g., median BHB and treatment duration) were assessed using Spearman’s correlation. Fisher’s exact test was used to analyze the relationship between 2 categorical variables in contingency tables (due to the small group sizes). A p < 0.05 was considered statistically significant.
Results
Study group characteristics
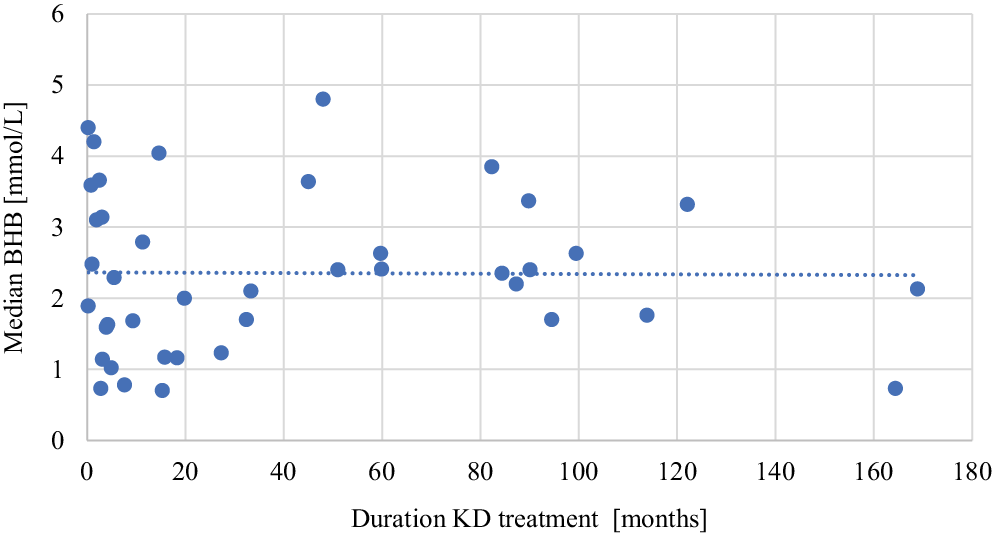
Forty-four patients with IEMs were included: 7 with various IEMs (group 1), 17 with MD (group 2), and 20 with GLUT1D or PDCD (group 3). Individual clinical and genetic data are presented in Supplementary Table 1. The median age at KD initiation in groups 1, 2, and 3 was 36.8, 52.7, and 53.1 months, respectively, and the median treatment duration was 4.9, 11.3, and 55.4 months, respectively. The total data for patients in each group are presented in Table 1. The vast majority of patients were treated with a classic KD; 6 patients received MAD, and an additional 14 patients used MCT oil. In groups 1, 2, and 3, the numbers of patients with a median BHB concentration below the therapeutic value were 3 (42.8%), 4 (23.5%), and 5 (25.0%), respectively. There was no correlation between the duration of KD treatment and the median BHB (r = −0.038, n = 41, p = 0.816; Figure 1).
Continuation vs discontinuation
Treatment with the KD is still ongoing in 20 patients: 42.8% with IEMs, 17.6% with MD, and 90% with GLUT1D and PDCD. The reasons for discontinuing treatment are listed in Table 2. Five patients (11.4%) discontinued the KD due to a lack of benefit after starting the KD, and the median duration of treatment was 3.9 (0.8–4.9) months. Adverse events were the reason for discontinuing the KD in 7 patients: 1 developed acute pancreatitis (after 14.6 months of the KD), 2 were found to have signs of fatty liver on ultrasound (after 3.9 and 18.3 months), and the others had troublesome GI symptoms (abdominal pain, vomiting); the median duration of the diet until discontinuation was 3 (0.2–11.3) months. Only 1 patient among those who discontinued treatment due to AEs did not benefit at all from the KD. During the KD treatment, 7 patients died, all due to disease progression; 5 were MD patients – the median survival in this group was 46.9 (4.5–219) months, and the duration of the KD treatment was 5.5 (1.4–168) months. Two patients with PDCD died at the ages of 37 and 24 months, after 33.3 and 4.2 months of the KD therapy, respectively. One PDCD patient, despite being treated with the KD from early infancy, had no benefit from the KD. Figure 2 shows the number of patients who continued the KD treatment in the relevant time intervals.
Benefits of treatment using KD
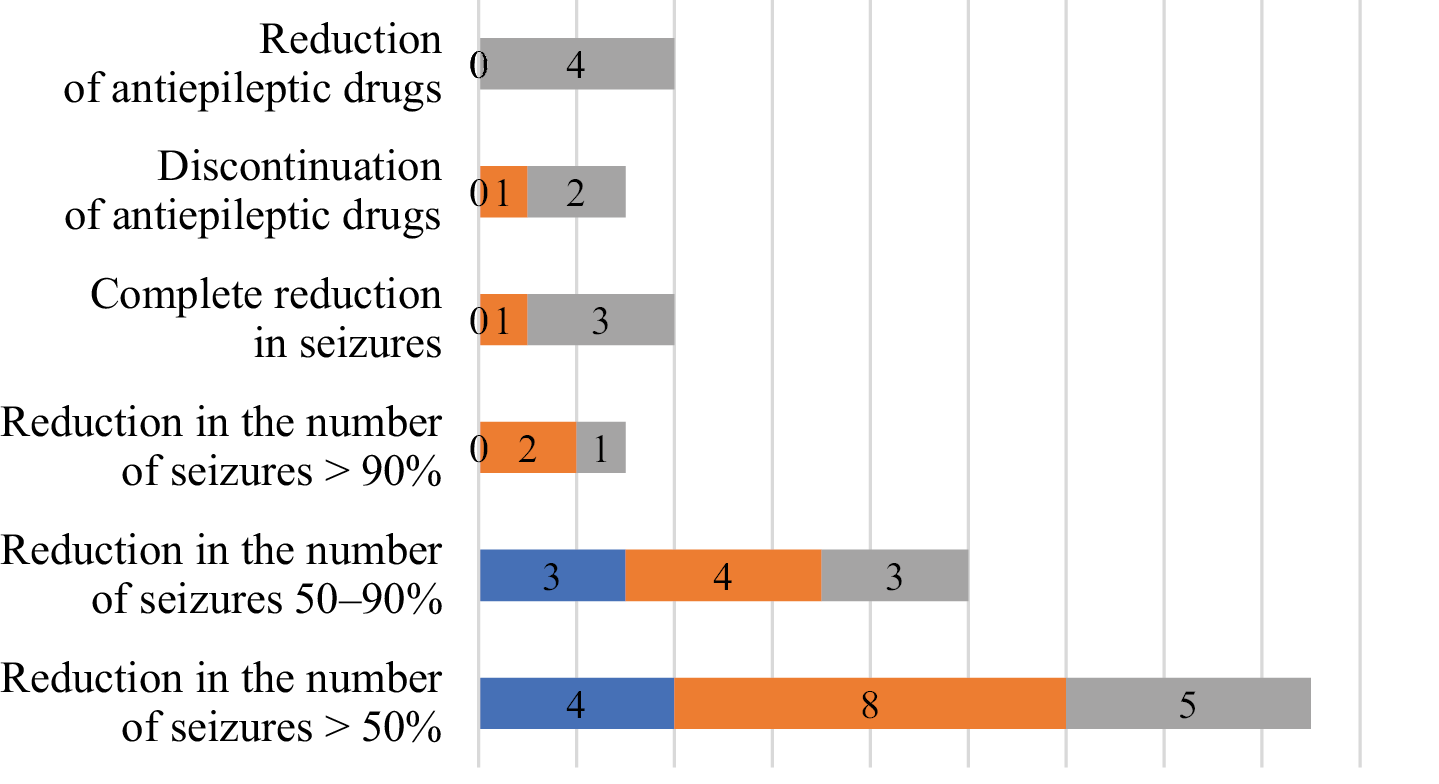
The entire group of 37 patients (84.0%) had some benefit from KD treatment. Among patients with seizures (n = 23), 17 (73.9%) experienced a reduction in the number of seizures (>50.0%), including a complete reduction in seizures in 4 (23.5%), a reduction of >90% in 3 (17.6%), and a reduction of 50–90% in another 10 (58.8%) patients. In addition, in 4 patients, the number of antiepileptic drugs (AEDs) was reduced, and in another 3 patients, they were completely discontinued (Figure 3). Antiepileptic drugs were reduced over a period of 6–12 months from the start of the KD.
Motor and functional outcomes
Among patients with abnormal muscle tone at baseline (n = 29), 8 (27.6%) showed improvement (reduced hypotonia or hypertonia) on clinical examination. Improved exercise tolerance was reported by caregivers in 21 of 41 evaluable patients (51.2%), although no objective exercise tests were applied. Improvement in ataxia was observed in 10 of 12 patients (83.3%) who had ataxia at baseline, and 9 of 15 (60.0%) showed a reduction in involuntary movements (e.g., chorea, dystonia). Caregivers of 8 out of 36 patients (22.2%) reported improvement in psychomotor or cognitive development, although standardized neuropsychological tests were not used. In 4 patients (3 with MD and 1 with GSD IIIa), a reduction or resolution of cardiomyopathy was observed during the KD. There were no significant differences in the pattern or magnitude of clinical benefit between the 3 diagnostic groups (Table 3).
Biochemical improvements
When patients were stratified according to median BHB (therapeutic ≥2 mmol/L vs non-therapeutic <2 mmol/L), there were no significant differences in clinical benefit (Table 4 and Supplementary Table 2). In 11 of 13 (84.6%) MD patients and in all PDCD patients, a decrease in lactic acid concentration was observed during the KD treatment. In patients with MD, the median difference in lactic acid concentration (at the start of treatment vs the last visit) was 1.07 mmol/L; in patients with PDCD, it was 1.5 mmol/L.
Adverse effects of KD
The most common abnormalities observed in laboratory tests were decreased serum free carnitine levels, hyperuricemia (>0.28 mmol/L), hypertriglyceridemia (>0.85 mmol/L (0–9 years) and >1.02 mmol/L (9–18 years)), hypercholesterolemia (>4.39 mmol/L), and metabolic acidosis (pH < 7.35 and BS <−5 mmol/L). In most cases, these abnormalities were transient. No symptomatic hypoglycemia was observed in any of the patients. Four patients had mild increases in alanine/aspartate aminotransferase (ALT/AST) activity: ALT (max 135 IU/L) and AST (max 242 IU/L), and in 1 patient, the diet was discontinued for this reason and due to signs of fatty liver and intolerance to the diet. In 1 patient (with GSDIIIa), transaminase activity decreased significantly after starting the KD (ALT: 65 vs 360 IU/L; AST: 66 vs 327 IU/L). No rhabdomyolysis was observed in any patient, and 2 patients had transiently elevated creatine kinase activity (355 and 483 IU/L), but these values were not persistent.
One PDCD patient had a prolonged QTc interval on routine ECG without clinical symptoms. Subsequent cardiology work-up (echocardiography, Holter monitoring, repeat ECGs) revealed no structural or rhythm abnormalities, and the KD was continued. Further ECGs remained normal.
Abdominal ultrasound in 5 patients revealed hyperechoic kidney changes, which resolved in 2 of them. Three MD patients had hyperechoic liver parenchyma suggestive of steatosis; in 2 of these, liver enzymes were moderately elevated. In 1 child, these abnormalities resolved after discontinuation of the KD, whereas in the other, they persisted and were most likely attributable to metabolic syndrome rather than to the KD alone.
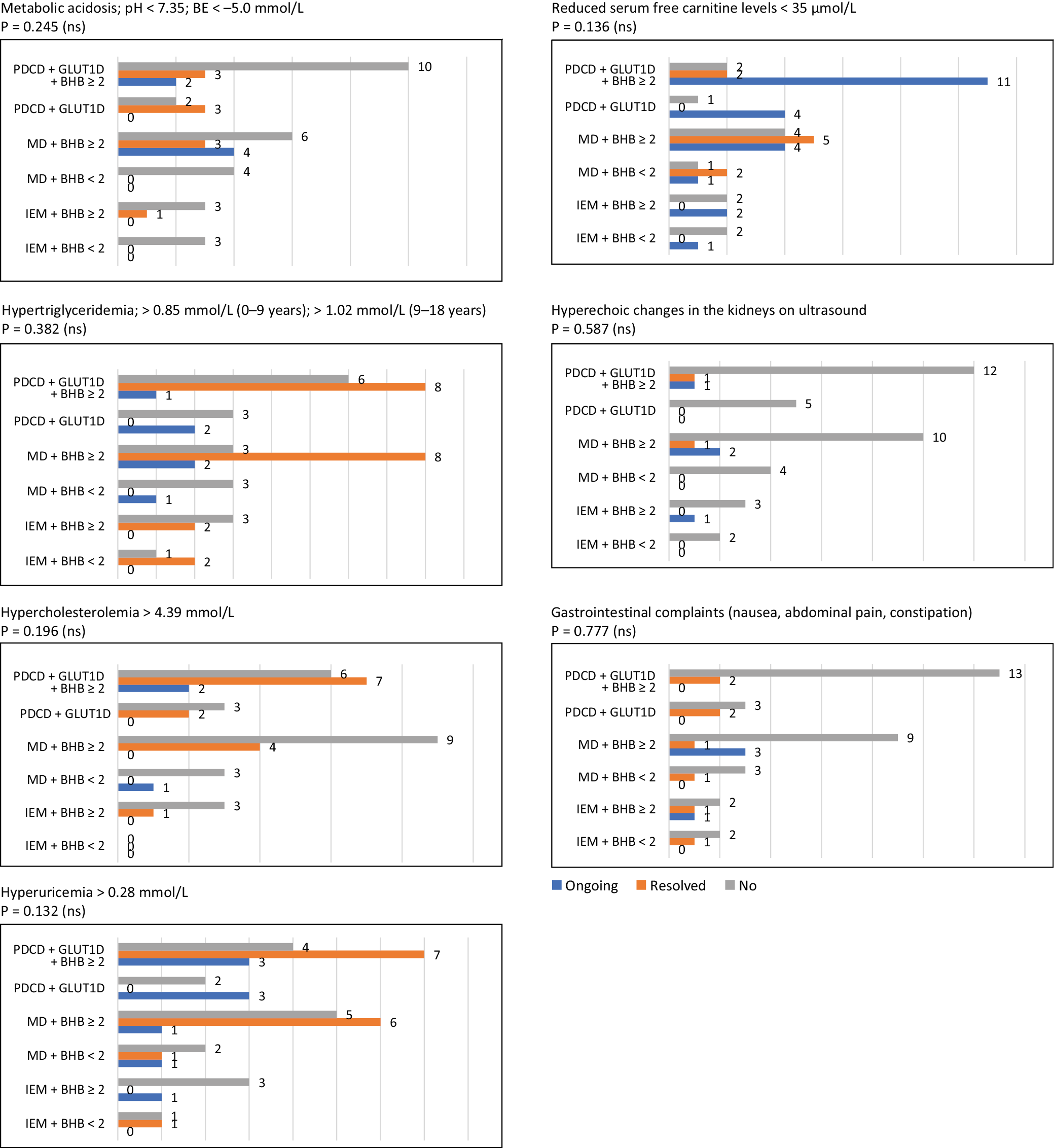
Gastrointestinal complaints (constipation, nausea, heartburn, abdominal pain) occurred in 10 patients. Constipation responded to standard treatment in all cases. In 4 patients, however, persistent GI symptoms led to KD discontinuation, despite very good seizure control. Comparing diagnostic groups and patients with median BHB ≥2 vs <2 mmol/L, no statistically significant differences in AE incidence were observed (Figure 4). Median maximum laboratory values by group and by BHB category are summarized in Supplementary Table 3 and Supplementary Table 4.
Growth and anthropometry
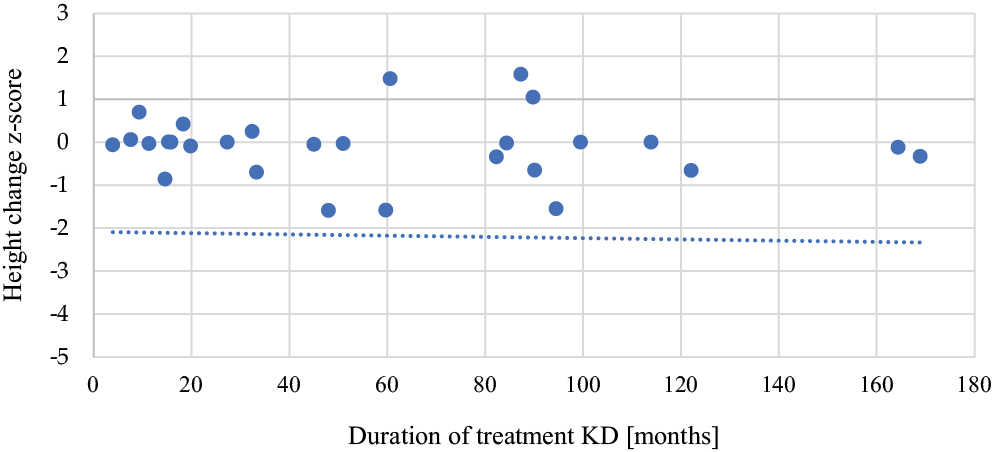
The median z-score of body weight difference (at the first and last visit) during KD treatment in the entire study group was 0.01 (−0.66–4.1). Three patients had z-score differences below −0.66, and 11 patients had z-score differences above 0.66. The median z-score of height during KD treatment was −0.005 (−1.59–1.58). Six patients had z-score differences below −0.66, and 3 patients had z-score differences above 0.66. There was no correlation between KD duration and height difference in patients treated with the KD (r = −0.194, n = 31, p = 0.295; Figure 5).
Discussion
The ketogenic diet has an established role in epilepsy management, including in very young children.16 Meta-analyses show that children on the KD are several times more likely to achieve ≥50% seizure reduction compared with standard care.5, 17 In our cohort, >70% of seizure patients achieved at least 50% reduction, and almost 1/4 became seizure-free, which is consistent with previous data. In GLUT1D and PDCD, the KD is considered first-line or essential therapy.1, 12, 18, 19 Literature data indicate high rates of seizure reduction and, to a lesser extent, improvements in movement disorders and cognition.20, 21, 22 There are many case reports describing the effectiveness of the KD in patients with PDHD, including the use of intravenous KD in newborns (2 case reports).23 In a serial magnetic resonance imaging (MRI) brain study of patients with PDCD, regression of lesions in the basal ganglia and clinical improvement were observed after starting the KD.24
In our study, 90% of PDCD/GLUT1D patients benefited from the KD, most often through seizure reduction, better muscle tone, fewer involuntary movements, and improved exercise tolerance. In PDCD, ketones bypass the defective pyruvate dehydrogenase complex, reduce glycolysis, and provide substrate for the Krebs cycle; this mechanism is reflected in the observed decrease in lactate levels. Nevertheless, some PDCD patients may not respond despite early initiation and adequate ketosis, as illustrated by 2 cases in our cohort. The diet is introduced as early as possible in these patients; there are even case reports of the use of intravenous KD in newborns (2 case reports).23 Treatment in this group of patients is recommended even for life (the longest observation is in a patient with PDCD – the KD for 14 years), which creates problems with compliance (1 patient with PDCD and 2 patients with GLUT1D were not reported in this publication due to non-compliance with dietary recommendations).
In the present study, there was no correlation between ketosis in patients and reported improvement, as well as the occurrence of AEs. However, in the literature, in both PDCD and GLUT1D patients, reported symptoms worsened after reduced ketosis, poor compliance, or discontinuation of treatment.13, 22, 25 In MD, the KD may improve mitochondrial function by enhancing oxidative metabolism, inducing mitochondrial biogenesis, reducing oxidative stress, and influencing mitochondrial dynamics.26, 27, 28, 29, 30 Several publications suggest that the KD can reduce seizures in MD patients and possibly improve other clinical outcomes.31, 32, 33 Several publications report resolution of status epilepticus after the introduction of the KD, but in some patients, this effect was only temporary, and in Alpers syndrome (an abnormal variant in the POLG gene), the KD does not improve survival.34, 35 Our patients with MD benefited from treatment using the KD primarily in terms of reducing epileptic seizures (80% showed improvement), but only 1 person managed to reduce the number of AEDs. This improvement is comparable to that in other groups.
Other benefits described in patients with MD treated with the KD include improvement in cognitive function,36 improvement in muscle tone and clinical status as assessed using the International Pediatric Mitochondrial Disease Scale (this study also included patients presented in the current paper).37 In our MD group, some had better muscle tone or functional status. Of particular interest is the partial or complete resolution of hypertrophic cardiomyopathy in 3 MD patients; such observations are supported by both clinical and animal data.38, 39, 40
Another group of IEMs in which treatment using the KD is effective is mitochondrial malate–aspartate shuttle (MAS) deficiency. Of the 13 individuals with MAS deficiency treated with the KD, 11 showed clinical improvement: a reduction in the number of epileptic seizures, improvements in muscle tone and motor development, better communication, and social interaction.41, 42 In the presented study, a patient with an abnormal variant in the SLC25A12 gene had no epileptic seizures at the time of KD initiation. After 8 months of the KD, a significant improvement in myelination was observed on MRI, and clinically, improved muscle tone and greater motor activity were observed – she was able to sit up on her own and, in the following months, crawl (before treatment, she would only lie down and had poor spontaneous activity). In addition, her AEDs were discontinued, and no seizures were observed (38 months without AEDs).
Nonketotic hyperglycinemia (NKH) is a severe neurometabolic disorder with epileptic encephalopathy and developmental delay from early infancy.43 Several case reports have shown that the KD can improve seizure control in patients with NKH and also reduce glycine concentrations in the body,44, 45, 46 including in brain tissue, although it does not normalize these concentrations.47 In our study, there was 1 child with NKH in whom we observed a significant reduction in epileptic seizures, but due to GI complaints, the parents decided to discontinue treatment.
The use of the KD in patients with glycogen storage diseases (GSD) is being studied primarily in GSD types IIIa, V, and VII. Ketone bodies are alternative energy sources for the brain, heart, and skeletal muscles, and by providing energy from fatty acids, they reduce the demand for glucose from glycogen. In addition, the KD has the potential to inhibit proteolysis in muscle, alleviate glucose depletion, and stimulate muscle regeneration or remodeling by enhancing satellite cell activation and differentiation. A low carbohydrate intake may prevent excessive glycogen accumulation in affected tissues (liver, muscles, heart).48
Data from a literature review showed that the introduction of a high-fat diet in patients with GSDIIIa resulted in decreased creatine kinase (CK) levels and reduced cardiac hypertrophy in children with GSDIIIa, but not in adult patients. Muscle strength, assessed with dynamometry, improved in only 1 patient, but subjective improvement in exercise tolerance and/or muscle strength was reported in 78% (14/18) of children with GSDIII and 50% (4/8) of adult patients.49 An additional benefit of the KD was a significant improvement in the maintenance of normoglycemia.48, 49, 50 It seems that patients with GSDIIIa may also benefit from a high-fat, high-protein diet to a similar degree as with the KD.48, 51
In our patient with GSDIIIa, after starting the KD, significant improvement in left ventricular hypertrophy, decreases in CK, aspartate transaminase (AST), and alanine transaminase (ALT) activity, and, above all, improved physical activity were observed. Also, in other muscle GSDs (types V and VII), studies suggest improvements in patient wellbeing and exercise tolerance.52, 53, 54 However, a large group of patients also drop out of the KD due to difficulties in adhering to dietary recommendations.55, 56 In addition to the abovementioned IEMs, the KD was used in individual cases of drug-resistant epilepsy in various IEMs, including succinic semialdehyde deficiency,57 arginine succinate lyase deficiency,58 various glycosylation disorders,59, 60, 61 and infantile Alexander disease.62 Treatment using the KD resulted in a reduction or even complete elimination of epileptic seizures in most patients.
In general, the spectrum and frequency of AEs in our cohort are similar to those reported in other KD studies.3 Most were mild or transient and manageable with relatively simple interventions. Gastrointestinal symptoms were the main cause of KD discontinuation, which is consistent with reports that up to 40% of KD-treated patients may experience significant GI complaints.3, 63
Limitations of the study
This study has several limitations. It is retrospective and may have missing data. The patient population is heterogeneous, encompassing different IEMs with diverse pathomechanisms and therapeutic targets for the KD, thereby limiting direct between-group comparisons. Many outcomes (e.g., exercise tolerance, developmental progress) were based on caregiver reports and clinical impressions rather than standardized assessments, which may introduce bias. Nonetheless, all patients were followed by the same team, which supports the internal consistency of evaluations. No significant differences in efficacy or AE incidence were detected between diagnostic subgroups, but small patient numbers limit statistical power.
Conclusions
The KD can be safely used in children with IEMs, and epilepsy is not the only indication for its implementation. The KD employs different mechanisms of action across IEMs, including providing ketones as an alternative energy source, modulating neurotransmission, and reducing specific metabolic loads (such as carbohydrates or glycine). In this cohort, the majority of patients (84%) benefited from the KD, most often through improved seizure control, as well as improvements in motor function, exercise tolerance, and, in some cases, cardiomyopathy and biochemical parameters. Side effects were usually mild and manageable; significant complications requiring discontinuation were relatively rare. However, GI complaints remain the most frequent reason for discontinuation or postponement of the KD, underscoring the importance of careful dietary counseling, monitoring, and individualized adjustments, particularly in the long-term treatment of IEM patients.
Supplementary data
The supplementary materials are available at https://doi.org/10.5281/zenodo.19365910. The package contains the following files:
Supplementary Table 1. Individual patient data.
Supplementary Table 2. Benefits of ketogenic diet treatment in individual groups depending on BHB concentration.
Supplementary Table 3. Median maximum laboratory results in each group.
Supplementary Table 4. Median laboratory results in patients with therapeutic and non-therapeutic level of BHB.
Data Availability Statement
The datasets supporting the findings of the current study are openly available in Zenodo at https://doi.org/10.5281/zenodo.19294948.
Consent for publication of personal information
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.