















Abstract
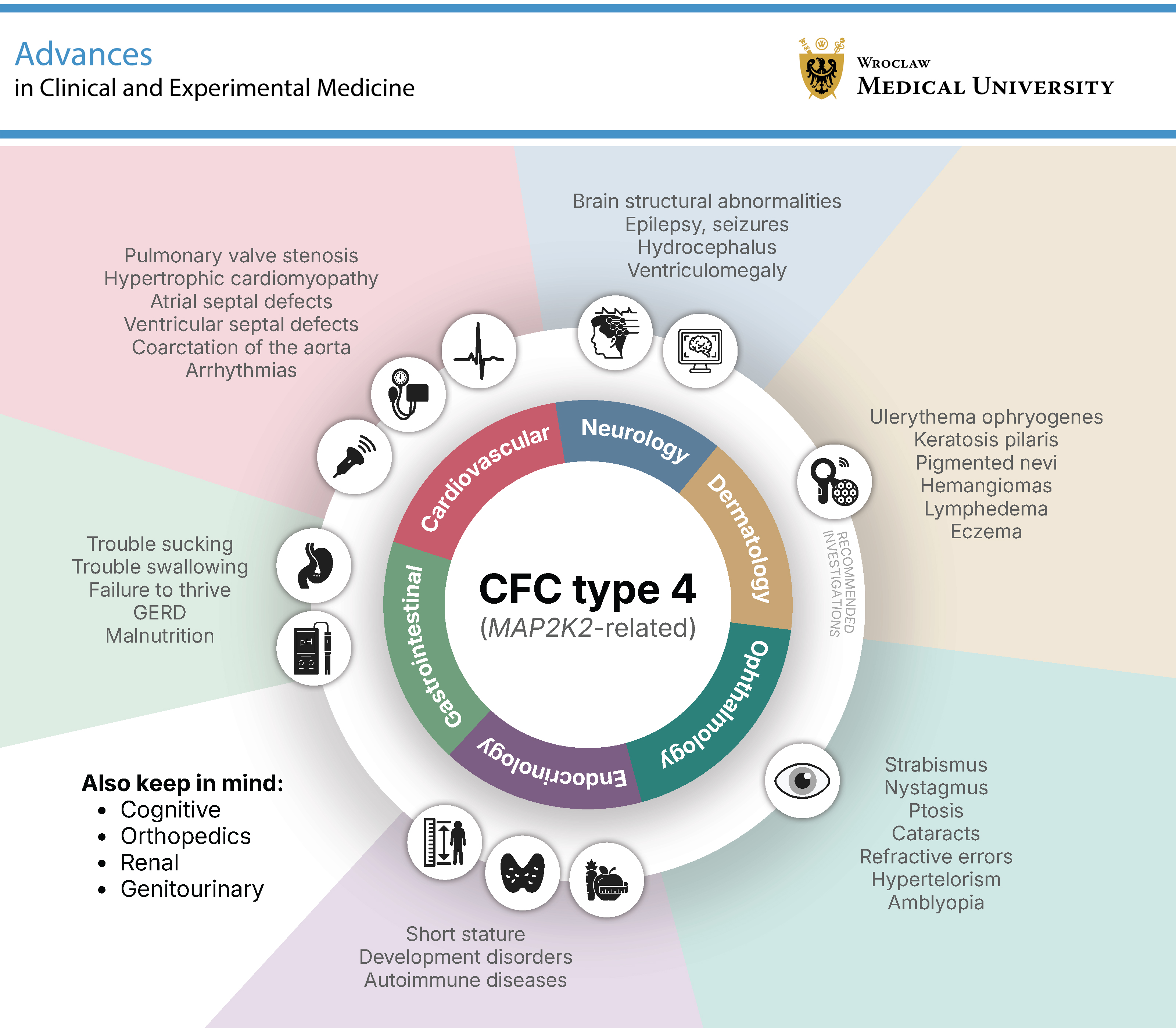
Cardiofaciocutaneous syndrome type 4 (CFC4) is a rare genetic condition caused by pathogenic variants in the MAP2K2 (MEK2) gene, part of the RAS/MAPK signaling pathway. While the broader phenotype of CFC syndrome has been well described, the features specific to this molecular subtype remain poorly defined due to the limited number of cases and underreporting. We conducted a structured analysis of all available literature on CFC4, focusing on organ-specific manifestations, including cardiac, craniofacial, neurological, integumentary, and gastrointestinal features, as well as developmental outcomes, treatment approaches, imaging findings, and behavioral profiles. Cardiofaciocutaneous syndrome type 4 is associated with a recognizable but variable phenotype. Pulmonary valve stenosis and atrial septal defects (ASDs) are the most common cardiac anomalies. Neurological involvement is nearly universal, often presenting as hypotonia and motor delay, with intellectual disability in a subset of cases. Distinctive craniofacial features and ectodermal abnormalities support clinical recognition. Feeding difficulties, sensory integration disorders, and behavioral challenges are frequently observed. Brain magnetic resonance imaging (MRI) abnormalities such as ventriculomegaly and corpus callosum hypoplasia are also relatively frequent. Notably, some individuals with CFC4 exhibit relatively mild phenotypes, with reports of independent functioning in adulthood and a history of familial transmission. In such cases, only mild learning difficulties were described. Better recognition and understanding of CFC4 require consistent and detailed reporting of new cases. To support this, we propose a concise clinical checklist to standardize case descriptions and support diagnosis.
Key words: RASopathies, MAP2K2, cardiofaciocutaneous syndrome, cardiofaciocutaneous syndrome type 4
Introduction
Cardiofaciocutaneous (CFC) syndrome is a rare developmental disorder that belongs to the group of RASopathies, conditions arising from germline mutations that dysregulate the RAS/MAPK signaling cascade. Most cases of CFC syndrome are attributed to variants in the BRAF gene; however, pathogenic variants in MAP2K1 (MEK1), KRAS, and MAP2K2 (MEK2) have also been implicated in the etiology of the disease. Notably, the same signaling alterations involved in these syndromes also contribute to tumorigenesis in a somatic context, further underscoring the biological significance of the RAS/MAPK pathway.1
Initially described in 1986, CFC syndrome was recognized as a distinct clinical entity based on the constellation of congenital heart defects, characteristic facial morphology, ectodermal abnormalities, and growth failure.2 The eventual discovery that Noonan syndrome (NS) and Costello syndrome (CS) are caused by pathogenic variants affecting the same signaling pathway helped explain the substantial overlap in clinical features with CFC.3 Cardiofaciocutaneous syndrome is most readily recognized by its characteristic craniofacial phenotype, which includes macrocephaly, a high anterior hairline, bitemporal narrowing, hypoplastic supraorbital ridges, and coarse or tightly curled hair. Ectodermal abnormalities are also prominent, particularly sparse or absent eyebrows, dry hyperkeratotic skin, and cutaneous vascular anomalies. Beyond the craniofacial and dermatologic hallmarks, CFC is a multisystem disorder affecting the cardiovascular, respiratory, neurological, gastrointestinal, and immune systems. Cardiovascular anomalies include pulmonary valve stenosis (PVS), atrial septal defects (ASDs), and hypertrophic cardiomyopathy (HCM). Neurological involvement ranges from hypotonia and gross motor delay to intellectual disability. Gastrointestinal manifestations, such as feeding difficulties and failure to thrive, are often present from infancy and may require prolonged nutritional support.4
Our understanding of CFC continues to expand. While genotype–phenotype correlations are beginning to emerge within and across RASopathies, they remain incomplete, especially for less common subtypes. Cardiofaciocutaneous syndrome type 4 (CFC4, OMIM:615280, Orpha:1340), caused by pathogenic variants in the MAP2K2 gene, is one of the rarest forms of the disorder. While the broader CFC phenotype has been relatively well characterized, data on CFC4 remain fragmented. The lack of comprehensive data on this subtype limits the development of tailored clinical management and effective treatments.
Objectives
This review examines the available literature on CFC4 and its phenotypic spectrum, based on published cases and comparative data. By synthesizing current evidence, we aim to outline the distinguishing features of CFC4 and propose a practical framework for its diagnosis and clinical management.
Methodology
We comprehensively reviewed the literature to identify reported cases of CFC4 associated with pathogenic variants in the MAP2K2 gene. We used the following search strategy across 4 major databases (PubMed, Embase, Scopus, and Web of Science) until March 2025: (“map2k2”[ti] OR “mek2”[ti] OR “cfc4”[ti] OR “cardiofaciocutaneous syndrome”[ti] OR “cfc syndrome”[ti] OR “cardio-facio-cutaneous”[ti]) NOT (“map2k1”[ti] OR “mek1”[ti] OR “mouse”[ti] OR “mice”[ti] OR “melanoma”[ti] OR “leukemia”[ti] OR “carcinoma”[ti] OR “sarcoma”[ti] OR “cancer”[ti]). This search yielded 289 records from Embase, 244 from PubMed, 257 from Scopus, and 326 from Web of Science. After removing duplicates, 337 unique articles remained. The literature search, screening, and study selection stages were conducted by a single reviewer (A.Ś.). Titles and abstracts were screened, and full texts of potentially eligible studies were reviewed.
Studies were included if they reported individual cases with genetically confirmed pathogenic variants in MAP2K2 consistent with a diagnosis of CFC4. Following full-text evaluation, 21 articles published between 2008 and 2023 met the inclusion criteria, comprising a total of 39 reported cases. After excluding 3 cases due to incomplete clinical data (originating from large cohorts without individual-level phenotypic information), the final dataset included 36 individual cases.5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22 A complete list of all reported cases is provided in Supplementary Table 1. No cases overlapped between the included studies.
Concurrently, we conducted a complementary literature review to obtain additional data on CFC4 phenotypic features, incorporating findings from larger cohort studies and broader reports on RASopathies that included patients with MAP2K2 variants. The search and study selection for this complementary review were conducted by 2 reviewers (H.K. and A.Ś.).
Results
Cohort summary
Our review of the literature identified 20 male and 16 female patients with CFC4. The mean age at the time of reporting was 18.61 years (range: 0.17–78 years), and the median age was 7 years. Of these, 3 patients died in infancy due to respiratory failure at the ages of 2, 3, and 9 months, respectively.5, 10, 12 A summary of the key clinical features extracted from previously reported cases identified in our literature search is presented in Table 1, Table 2. The following sections provide a detailed, system-by-system description of CFC4 phenotypic findings based on individual case reports. Given the limited number of cases, we integrated our findings with data from broader literature reports on CFC and related RASopathies that included patients with MAP2K2 variants within larger cohorts. Management strategies tailored to each organ system are presented in the Management subsection of the Discussion.
Prenatal phenotype
Polyhydramnios was the most common prenatal finding in CFC4, reported in 12 of 20 cases with available data (60.0%). Other prenatal abnormalities, including omphalocele, maternal diabetes, increased nuchal translucency (NT), hepatomegaly, unilateral hydronephrosis, intrauterine demise of a co-twin, premature uterine contractions, and cystic hygroma colli, were much less common and typically occurred as isolated findings.
Although RASopathies occur in approx. 1 in 1,000 live births, prenatal diagnosis remains challenging due to their largely non-specific fetal phenotype.23 Data from Dutch and Chinese cohorts confirm that increased NT is the most frequent ultrasound finding, often detected in the 1st trimester, with a median NT of 5.0 mm and a substantial proportion of cases presenting with NT ≥ 6 mm.24, 25 The diagnostic yield for pathogenic variants in cases referred for genetic testing based on suggestive ultrasound findings is only about 9–14%.24, 26 Templin et al. reported that the constellation of prenatal features in CFC consists of macrocephaly, macrosomia, and polyhydramnios, frequently accompanied by shortened femora (similar to that observed in CS).26 Among the gestational abnormalities described, polyhydramnios was the most common overall (52%), followed by urinary tract anomalies (47%), including renal cysts and pyelectasis.26 According to Scott et al., the recommended approach for prenatal genetic testing for RASopathies is to perform chromosomal microarray analysis in fetuses with increased NT or other anomalies, followed by RASopathy gene panel or exome sequencing if the microarray is normal, especially when NT ≥ 5.0 mm or NT ≥ 3.5 mm with additional findings, or when lymphatic dysplasia or congenital heart disease is present.27 Findings from a retrospective questionnaire study of CFC patients showed that the prevalence of gestational diabetes (7%) and intrapartum infections (14%) were similar to those in the general population. In comparison, hyperemesis gravidarum (11.6% vs 3%) and pregnancy-induced hypertension (14% vs 5%) occurred at higher rates than in the general population.28 Moreover, the detection of pregnancy complications increased across trimesters (10% in the 1st, 60% in the 2nd, 79% in the 3rd), and 37% of pregnancies underwent invasive prenatal diagnosis.28
Early postnatal phenotype
Among 19 CFC4 patients with available data, preterm delivery occurred in 7 cases (36.8%), with the earliest birth recorded at 27 weeks of gestation. Birth parameters, standardized for sex and gestational age using the Fenton and Kim growth charts,26 revealed a mean birth weight z-score of +0.34 standard deviation (SD) (n = 17). Only 2 infants exceeded +2 SD, while 2 were below −2 SD, indicating no consistent pattern of growth restriction or macrosomia. Head circumference data were available for 14 patients, with a mean z-score of +1.67 SD (range: from −3.5 to +5.5). Macrocephaly (z-score > +2 SD) was confirmed in 9 of 14 patients (64.3%). Respiratory compromise was a notable early postnatal finding: 5 neonates (26.3%) developed respiratory distress, all requiring neonatal intensive care unit (NICU) admission.
Jelin et al. reported that infants with CFC have a markedly elevated rate of preterm birth, with a mean gestational age at delivery of approx. 35 weeks and fewer than 15% reaching term; the majority experience a complicated neonatal course, including prolonged hospitalization (mean length of stay >1 month), frequent need for feeding tube placement (57%), and a substantial proportion requiring intubation and respiratory support (26%).28 Early postnatal complications include chylothorax, gross edema, hyperbilirubinemia, hypoglycemia, and arrhythmias.25
Craniofacial and ectodermal phenotype, dermatologic manifestations
Craniofacial and ectodermal features in genetically confirmed CFC4 cases are shown in Table 1. The most consistently observed craniofacial features included macrocephaly (81.8%), a high forehead, bitemporal narrowing, and a tall chin. Ocular findings were frequent, with down-slanting palpebral fissures reported in 69.4% of cases, followed by hypertelorism, hypoplastic supraorbital ridges, and epicanthus. Less common but notable features included bilateral ptosis, strabismus, exophthalmos, congenital cataracts, and optic nerve hypoplasia. Auricular anomalies, particularly low-set and posteriorly rotated ears, were characteristic, while nasal features often comprised a depressed bridge with a bulbous tip. Oral findings included a long philtrum, thin upper lip vermilion, high-arched palate, and, in some reports, micrognathia or anterior open bite. Ectodermal abnormalities were highly prevalent and often supportive of the diagnosis. Sparse eyebrows (84.8%) and sparse eyelashes (81.8%) were reported in most patients, frequently accompanied by brittle, curly, or sparse scalp hair. Cutaneous involvement included keratosis pilaris, palmoplantar hyperkeratosis, and infantile hemangiomas, most commonly in the occipital region. Other less frequent but relevant features included multiple melanocytic nevi, café-au-lait macules, dermatitis, hyperextensible skin, and nail dysplasia.
The craniofacial phenotype observed in CFC4 largely mirrors the broader CFC pattern described in larger mixed-genotype cohorts.29, 30, 31 Typical traits (macrocephaly, prominent forehead, bitemporal narrowing, down-slanting palpebral fissures, and low-set, posteriorly rotated ears) form a recognizable gestalt, often apparent in early childhood. Collectively, previous studies have highlighted the high prevalence of skin manifestations in CFC.32 Keratosis pilaris is reported in most patients (82%), with generalized involvement in nearly ¼ of cases.32 Ulerythema ophryogenes occurs in approx. 44% of cases and typically affects the eyebrows, cheeks, and helices.32 Palmoplantar hyperkeratosis is noted in 27% of individuals, presenting as bilateral, painful keratotic plaques over pressure points.32 Multiple melanocytic naevi (>50) are observed in 29% of patients and are widely distributed without photodistribution preference.32 Prior studies report that hair abnormalities occur in most CFC patients, including wavy or curly scalp hair (69%), slow-growing hair (58%), temporal alopecia (59%), and sparse or absent eyebrows (73%).32
In this cohort, only 2 individuals carried MAP2K2 variants, yet both exhibited the characteristic ectodermal profile, including wavy or curly scalp hair, markedly slow hair growth, and sparse or absent eyebrows.32 One patient additionally demonstrated sparse or absent eyelashes and temporal alopecia. Prior analyses have further shown that hair abnormalities are significantly more frequent in individuals with BRAF variants compared with those with MAP2K1 or MAP2K2 variants (97% vs 60%).32 However, no single hair phenotype is distinguishing.
Cardiac manifestations
In our review, 58.8% of individuals with CFC4 had a cardiac defect, most commonly PVS, observed in 41.2% of individuals, with nearly 1/3 requiring surgical intervention. Atrial septal defects were the 2nd most common, affecting approx. 1/5 of patients. Other findings were reported only sporadically, including ventricular septal defects (VSDs), cardiomyopathy, cardiac arrhythmias, bicuspid aortic valve, patent foramen ovale, and pulmonary hypertension. Cardiomyopathy was rare (5.9%), which may reflect the young age of many patients, incomplete follow-up, or a genuinely lower prevalence in this genotype.
Cardiac involvement is a hallmark of CFC syndrome, historically reported in 60–80% of cases.4, 29, 33, 34 While many anomalies are detected neonatally, some, particularly HCM and arrhythmias, may appear later in childhood and carry substantial risk for heart failure or sudden cardiac death.35 When comparing cardiac manifestations across genotypes, several patterns emerge. Hypertrophic cardiomyopathy, PVS, and ASD were all reported more frequently in individuals with BRAF variants than in those with MAP2K1 or MAP2K2 variants; however, only the difference in PVS reached statistical significance (50% vs 37%, p = 0.000363).36 Conversely, VSDs were more common in the MAP2K1/MAP2K2 cohort compared with BRAF (19% vs 11%).36 These findings align with previous observations suggesting a lower overall burden of structural heart disease in CFC4. Armour et al. reported cardiac involvement in approx. 50% of MAP2K2-positive individuals, compared with 72–75% in those with BRAF or MAP2K1 variants.33 However, that study included only 2 MAP2K2 patients, and the sole cardiac abnormality described was HCM, with no documented cases of PVS, VSDs, or ASDs.33 Feng et al. observed higher rates of PVS and ASDs in a cohort of Chinese patients with MAP2K1/2 variants compared with those carrying BRAF variants.34 In the same study, HCM was notably less common in the MAP2K1/2 group than in the BRAF group.34 The authors also reported a high prevalence (22%) of PVS in MAP2K1/2 cases, with no HCM observed in this subgroup.34
Gastrointestinal manifestations and feeding difficulties
In our review of CFC4 cases, feeding and gastrointestinal complications were common. Failure to thrive was reported in 41.9% of patients, gastroesophageal reflux disease (GERD) in 25%, and sucking and swallowing dysfunction in 20%. Approximately 25.8% of individuals required tube feeding during infancy or early childhood. Pyloric stenosis was identified in 12.5% of cases and, when present, required surgical intervention in 75% of affected individuals. Other gastrointestinal findings included abdominal hernias and non-specific feeding difficulties.
These findings align with previous reports of gastrointestinal involvement in CFC syndrome. Feeding difficulties are among the earliest and most persistent challenges in affected patients, described in up to 95% of individuals.34 Problems typically begin in the neonatal period and include poor sucking, swallowing dysfunction, GERD, and oral aversion, often resulting in prolonged feeding times, frequent gagging, spitting, or complete food refusal.4, 30, 37 Oral hypersensitivity further complicates the transition to solid foods, as approx. 1/3 of patients never develop adequate chewing abilities.37 Feeding problems frequently lead to failure to thrive and may necessitate long-term nutritional support via nasogastric tube or gastrostomy tube.30, 33 Additional gastrointestinal features described in the literature include vomiting, dysmotility, constipation, and drooling, reported in up to 27% of patients, with 11% experiencing constant drooling.33, 37 Caregivers often describe mealtimes as stressful and prolonged (40–50 min) due to feeding refusal and marked selectivity for food texture and taste, while excessive hunger is reported in 42% of cases.37
Somatic growth
Given incomplete documentation across reports, sex- and age-adjusted z-scores were calculated based on available data. Growth assessments in our review showed a consistent trend toward short stature, with a mean weight z-score of −0.70 (n = 11; range: from −2.02 to +0.83) and a mean height z-score of −1.80 (n = 15; range: from −6.00 to +0.89) among CFC4 patients.
These findings are consistent with previous literature, which describes growth impairment as a core feature of CFC syndrome. Many individuals have normal weight and length at birth, with growth velocity often declining during infancy. This results in weight and stature typically falling below the 5th percentile while head circumference remains preserved, leading to relative macrocephaly.4 Among RASopathies, CS and CFC are notable for their association with musculoskeletal malformations.38 It remains unclear whether these malformations result directly from the disease’s pathomechanism or are secondary to growth hormone (GH) deficiency.
Neurodevelopment, cognition, and behavior
Developmental delay emerged as a consistent and defining feature across reported CFC4 cases. Intellectual disability or learning difficulties were reported in 65.6% of individuals, with severity ranging from mild to profound. Hypotonia was present in 40.0% of cases, while motor delay was reported in 37.9%.
Developmental delay is also a defining feature of CFC syndrome more broadly, with severity ranging from mild motor impairment to profound intellectual disability.29 Hypotonia and delayed motor development (particularly affecting gross motor milestones) are among the earliest and most consistent manifestations, often present from infancy.4, 39, 40 Independent walking may not be achieved until the age of 3 years or later.41 Fine motor skills are usually less delayed but still impaired. Speech and language development are often severely affected, with first words typically emerging after the age of 2, and some individuals remaining nonverbal.41 Expressive language is consistently weaker than receptive abilities,39 and independent, fluent verbal communication is rare, reported in fewer than 10% of cases.42 Adaptive functioning is frequently compromised, particularly in mobility and daily living skills. Standardized functional assessments show that individuals with CFC typically score 3–5 SD below age-matched peers in mobility, self-care, and social cognition,40 with only modest improvement over time. Sensory processing dysfunction has been reported in over 85% of patients, characterized by sensation-seeking behaviors, tactile hypersensitivity, and patterns of low muscle tone or reduced strength.42 These difficulties often co-occur with emotional and behavioral issues, including distractibility, irritability, and social withdrawal, suggesting shared neurodevelopmental mechanisms.
Neuropsychiatric and behavioral features, including irritability, anxiety, obsessive-compulsive behaviors, and autistic traits, are also described and may substantially impair quality of life.29 Self-injurious, aggressive/destructive, and stereotyped behaviors are common in CFC, with the highest burden in individuals with BRAF and MAP2K1 variants and during adolescence.43 The MAP2K2 cohort consistently exhibited the lowest frequency of these behaviors.43 Self-injurious behaviors are particularly prevalent, reported in more than 90% of patients, most commonly head-hitting, self-scratching, and teeth grinding.43 Aggressive or destructive behaviors are also frequent, affecting approx. 2/3 of individuals (69%), and typically manifest as pushing, hitting, or grabbing others.43 Stereotyped behaviors (including yelling and screaming, waving or shaking of arms, repetitive body movements, and hand clapping) are highly prevalent in patients with CFC, affecting approx. 95% of individuals.43 Epilepsy, intellectual disability, reduced adaptive functioning, chronic pain (including abdominal and musculoskeletal pain), limited expressive communication, and sleep disturbances all contribute to an increased likelihood of challenging behaviors in CFC.43 Notably, although neurocognitive impairment is common, a subset of individuals with CFC has been reported to exhibit normal intellectual functioning.29 Familial transmission was described in several cases,6, 8, 22 suggesting that a subset of individuals with CFC4 may present with milder phenotypes compatible with independent adult functioning.
Neurological manifestations
Across reported CFC4 cases, seizures were infrequent, occurring in 6.9% of cases, and when present, were generally mild in severity. Brain imaging data were limited, as magnetic resonance imaging (MRI) was not consistently performed. Among patients with available imaging (n = 18), ventriculomegaly was the most frequent finding (42.1%), followed by corpus callosum hypoplasia (22.2%) and cerebellar hypoplasia (16.7%). Additional isolated findings included craniosynostosis with type I Chiari malformation,16 cerebral atrophy,11 and periventricular leukomalacia.13
The broader literature confirms that seizure disorders are reported in up to 55–65% of individuals with CFC. However, both prevalence and severity appear lower in CFC4 (30%) compared with BRAF- or MAP2K1-related cases.29, 44 When present, seizures most commonly manifest as generalized tonic-clonic (34%) and focal (33%), followed by absence (25%), myoclonic (12%), and infantile spasms (10–12%).44 Among CFC patients, structural brain abnormalities are likely underrecognized, as brain imaging has been performed in only 38–50% of patients in published cohorts.45 Anomalies reported in the literature include ventriculomegaly (43.9%), hydrocephalus (24.2%), cortical atrophy (12.1%), and prominent perivascular spaces (10.6%), with less frequent findings such as corpus callosum hypoplasia, cerebellar hypoplasia, delayed myelination, and brainstem atrophy.45
Immune system
In our CFC4 cohort, the available data on immune manifestations were insufficient to support meaningful conclusions, likely reflecting inconsistent clinical reporting and the absence of systematic immunologic screening. Emerging evidence suggests that RASopathies may be associated with autoimmunity, particularly autoimmune thyroiditis. Elevated anti-thyroid peroxidase antibodies have been reported in up to 52% of affected individuals, highlighting the value of immunological screening during follow-up.46 This predisposition may stem from MAP2K2-mediated overactivation of the RAS/MAPK pathway. The ERK cascade is crucial for the early development of B and T cells.47 Preclinical studies suggest that dysregulation of the RAS/MAPK pathway can drive autoimmune-like phenotypes through multiple immune mechanisms. However, the clinical significance of these findings remains unclear due to a lack of corresponding human data.48, 49
The most recent study by Di Majo et al.,50 involving 56 patients with CFC syndrome, provides new insights into mutations in the RAS/ERK pathway and their impact on immune function. The authors report an association between CFC and lymphopenia, hypogammaglobulinemia, and increased susceptibility to infections. However, these findings require further validation through additional research. The authors propose that CFC may represent a form of “syndromic immunodeficiency.”
Renal and genitourinary manifestations
There were insufficient data on genitourinary manifestations in our cohort to draw conclusions specific to CFC4. However, renal and other genitourinary abnormalities have been reported in up to 33% of individuals with CFC.4 Cryptorchidism and renal or bladder malformations appear relatively common, while additional findings such as neonatal renal enlargement, prenatal hydronephrosis, duplex collecting systems, enlarged kidneys, and vesicoureteral reflux have also been described. Notably, nocturnal enuresis has been reported in patients up to 18 years of age. In an analysis limited to individuals with MAP2K2 variants, all affected males (2/2) had cryptorchidism, and 1 also exhibited a renal or bladder abnormality.33
Ophthalmologic manifestations
In our cohort, no clear pattern suggesting a predominance of specific ophthalmologic abnormalities was observed. Bilateral ptosis was documented in 6 patients, strabismus in 3, and cataracts in 2. Single cases of nystagmus, optic nerve hypoplasia, astigmatism, and diplopia were also reported.
Ophthalmologic findings most frequently reported in CFC include strabismus, ptosis, refractive errors, and optic nerve hypoplasia. Other ophthalmologic findings such as myopia, astigmatism, nystagmus, and amblyopia are also observed. Retinal dystrophy has been reported in some patients, although its prevalence and natural history are not yet fully understood.51 Crincoli et al. concluded that refractive profiles in CFC patients differ significantly depending on the underlying genetic variant.52 Their analysis demonstrated that individuals with BRAF variants more frequently exhibited emmetropia or only mild refractive defects, as well as higher rates of astigmatism >1.5 D. In contrast, patients carrying KRAS, MAP2K1, or MAP2K2 variants showed a markedly increased prevalence of high myopia (>6 D) and hyperopia. Notably, among this group, only 1 CFC4 patient was included, a 9-year-old boy with a marked refractive error, with high myopia in both eyes (−8.5 D in the right eye and −7.0 D in the left), accompanied by nystagmus, exotropia, and bilateral optic nerve pallor. Despite the severity of the refractive defect, the patient achieved normal best-corrected visual acuity of 20/20 in both eyes.52
Hearing
Hearing loss was reported in 2 patients in our cohort. Although hearing impairment, including sensorineural hearing loss, has been described in individuals with CFC, it is not considered among the most common or characteristic clinical features. Many affected children experience recurrent otitis media and often have narrow external auditory canals, with a substantial proportion requiring the placement of pressure-equalization tubes.4
Malignancies
Three cases of malignancy have been reported in the CFC4 cohort, occurring across a wide age range and involving diverse tumor types: orbital meningioma associated with extranodal Rosai–Dorfman disease at 16 years, cervical cancer at 27 years, and diffuse large B-cell lymphoma at 70 years.14, 19, 22, 53
The reported incidence of malignancy in CFC4 appears to be very low and primarily involving hematologic malignancies in early childhood, with no evidence of a unique cancer profile. In a broader review of CFC cases published from 1937 to 2010, Kratz et al. identified 8 malignancies (3.5%) among 226 CFC patients.53 These included 4 cases of acute lymphoblastic leukemia (median age, 5 years), 2 cases of non-Hodgkin lymphoma, as well as 1 case of hepatoblastoma and 1 of rhabdomyosarcoma. Although the number of cancers observed in CFC remains too small to permit definitive conclusions regarding risk and prevalence, the spectrum of malignancies reported in CFC overlaps with that seen in other RASopathies, including NS, CS, and NS with multiple lentigines. In another survey by Bess et al., including 690 individuals with BRAF, MAP2K1, or MAP2K2 variants, 11 cases of cancer were reported.54 Six of these were associated with known pathogenic variants. The most frequently observed malignancies were acute lymphoblastic leukemia and non-Hodgkin lymphoma. Isolated cases included hepatoblastoma, rhabdomyosarcoma, and oligodendroglial leptomeningeal tumors. Both the overall incidence of cancer and the specific incidence of acute lymphoblastic leukemia were significantly higher than in the general population. Among CFC4 cases specifically, malignancy remains exceedingly rare, with only isolated reports and no clear evidence of a distinct cancer spectrum.
Discussion
Our analysis refines the current clinical understanding of CFC4. In the following discussion, we outline diagnostic considerations, management strategies, and propose a standardized framework for future case reporting (Table 3).
Diagnosis and reporting
Cardiofaciocutaneous syndrome is suspected based on distinctive clinical features and confirmed by molecular genetic testing.30 In the context of CFC4, several craniofacial features are particularly suggestive, including a high forehead, bitemporal narrowing, hypoplastic supraorbital ridges, down-slanting palpebral fissures, a broad nasal bridge, and low-set, posteriorly rotated ears with thickened helices. These findings are frequently accompanied by congenital heart defects, most commonly PVS or cardiomyopathy, as well as significant developmental delay, hypotonia, and gastrointestinal dysfunction, often manifesting as failure to thrive, poor oral feeding, or long-term tube feeding requirements.4, 29
Molecular confirmation remains the gold standard for diagnosis, with next-generation sequencing (NGS)-based multigene panels covering RASopathy-associated genes providing the highest diagnostic yield, detecting pathogenic variants in approx. 70–90% of clinically suspected cases. When panel testing is inconclusive, targeted single-gene sequencing or chromosomal microarray analysis may be considered to detect copy number variants.29 Early and precise molecular diagnosis is crucial for establishing the clinical subtype and guiding anticipatory management and genetic counseling.
The differential diagnosis of CFC4 overlaps with other RASopathies, particularly NS, Noonan syndrome with multiple lentigines (NSML), CS, and neurofibromatosis type 1 (NF1), but can also include Legius syndrome, central conducting lymphatic anomaly, capillary malformation–arteriovenous malformation syndrome, and neurodevelopmental disorders such as SYNGAP1-related encephalopathy.29
Given the rarity of CFC4 and the variability of its presentation, consistent and detailed reporting of new cases is essential to refine our understanding of this condition. We recommend the use of a standardized reporting framework that captures the full clinical, genetic, and developmental spectrum of CFC4. Reports should include a structured clinical phenotype organized by system (particularly cardiac, craniofacial, neurologic, dermatologic, and gastrointestinal features), as well as prenatal history and detailed birth parameters such as gestational age, Apgar scores, neonatal complications, and admission to the NICU when applicable. Developmental outcomes should be described with attention to both motor milestones and cognitive or social functioning, as well as imaging results, including brain MRI or computed tomography (CT), if performed. Genetic variants should be reported using Human Genome Variation Society (HGVS) nomenclature, together with information on the methods used and any limitations of testing. Longitudinal clinical data are equally important: current growth parameters, relevant treatments, surgical interventions, and educational progress enrich the understanding of disease impact. When possible, family history should be included, noting the presence or absence of similar phenotypic traits. Such harmonized case descriptions will improve phenotypic delineation, facilitate genotype–phenotype correlations, and support the development of evidence-based management guidelines for patients with CFC4.
Management
Cardiofaciocutaneous syndrome type 4 presents with a broad phenotypic spectrum, requiring coordinated, multidisciplinary care throughout life. Currently, no curative therapy exists, and management remains symptomatic and anticipatory. Experimental strategies, such as MEK inhibitors and antisense oligonucleotide (ASO) therapies, are under early clinical investigation (Walczuk, personal communication). Below, we summarize established management recommendations4, 30 for major organ systems and highlight promising therapeutic directions. It should be noted that some clinical recommendations and therapeutic options are based on data from all CFC subtypes or from other RASopathies.
Cardiology
Echocardiography is recommended at diagnosis and at regular intervals (every 12–24 months in childhood, with individualized follow-up thereafter).30 Pulmonary valve stenosis is the most common defect, typically treated with cardiac catheterization and balloon valvuloplasty.55 To prevent progression, the American Heart Association (AHA) and the American College of Cardiology (ACC) recommend prompt balloon valvuloplasty after diagnosis, even in asymptomatic patients.56 For HCM, propranolol, introduced by Shand et al.57 and later optimized to 6 mg/kg by Östman-Smith,58 remains the first-line therapy, significantly improving survival in pediatric patients. Calcium channel blockers are generally avoided due to increased mortality risk. Severe or resistant cases may require surgical myomectomy or, rarely, heart transplantation.
Neurology and development
Currently, rehabilitative interventions remain the only established treatment for cognitive impairment. Individualized programs may include specialized education, speech therapy, occupational therapy, physical therapy, and caregiver support. Evidence from related conditions suggests that sensory-based therapies, functional communication training, sign language, and naturalistic teaching approaches can benefit children and their families.42, 59
Epilepsy, though less common in CFC4, may manifest in patients with CFC syndrome broadly as tonic-clonic seizures, focal seizures, or absence seizures. Detailed monitoring of seizure frequency, duration, type, and response to medication is essential, with severity assessed using tools such as E-CHESS.59 Management usually requires polytherapy, commonly with sodium channel blockers (zonisamide, lacosamide) and benzodiazepines (diazepam, clonazepam), while levetiracetam has limited efficacy in CFC syndrome and frequent adverse effects. Non-pharmacological options, including epilepsy surgery, vagus nerve stimulation, the ketogenic diet, or callosotomy, have been beneficial in select cases.59, 60, 61
Management of hypotonia primarily relies on standard rehabilitation programs and physical therapy.30 Coenzyme Q10 supplementation has been associated with clinical improvement in isolated reports62; however, these observations remain anecdotal, with no reproducible data confirming its efficacy.
Another important aspect of RASopathies that requires clinical management is the increased prevalence of psychiatric disorders.63, 64 The authors recommend routine evaluation of sleep disturbances as part of standard care, emphasizing that psychiatric assessments should be conducted regularly to facilitate early detection and initiation of appropriate therapy, whether psychotherapeutic or pharmacological. A study by Geoffray et al.65 showed that patients with CFC exhibited impairments in reciprocal social interaction, restricted repetitive behaviors, reduced shared enjoyment, anxiety, tantrums, and overactivity. Their results suggest that treatment insights gained from studying one RASopathy may apply to other types, despite differences in molecular mechanisms.
Endocrinology, immunology, and growth
Currently, the main indication for GH therapy is a confirmed GH deficiency. However, GH affects not only linear growth but also psychomotor development, bone mineral density, lipid and carbohydrate metabolism, body composition, and overall wellbeing.66 Therefore, in some countries, NS is considered a valid indication for GH therapy regardless of GH levels, with proven beneficial outcomes.67 Although CFC syndrome itself is not currently approved as an indication for GH therapy, further research is warranted, as evidence from related RASopathies suggests potentially significant benefits. Beyond GH therapy, regular monitoring of growth parameters (height and weight) throughout infancy and childhood is recommended, along with assessment of parathyroid hormone, calcitonin, and vitamin D levels to enable early detection and management of skeletal abnormalities.68
An emerging therapeutic option for growth failure in RASopathies is C-type natriuretic peptide (CNP) analogues, particularly vosoritide (clinical trial: NCT04219007). This U.S. Food and Drug Administration (FDA)-approved oligopeptide, currently used in the treatment of achondroplasia, binds to natriuretic peptide receptor type B and attenuates RAF signaling, thereby counteracting overactivation of fibroblast growth factor receptor 3 (FGFR3).69 In murine models with BRAF variants, CNP analogues increased body length and raised insulin-like growth factor 1 (IGF-1), insulin-like growth factor-binding protein 3 (IGFBP-3), and GH levels, suggesting potential benefit in CFC patients.70 However, this approach remains experimental and requires further research.
Elevated anti-thyroid peroxidase antibodies have been reported in up to 52% of affected individuals.46 Regular screening for autoimmune thyroiditis may therefore be considered.
Gastroenterology and feeding
The most frequently reported gastrointestinal complications in patients with CFC include oral aversion, GERD, and sucking and swallowing dysfunction. Placement of a gastrostomy tube is relatively common and is often required due to persistent feeding difficulties. First-line pharmacological treatment for GERD includes proton pump inhibitors. In more severe or treatment-resistant cases, surgical intervention, such as Nissen fundoplication, can be an effective option.71 Regular follow-up with a pediatric gastroenterologist is crucial for monitoring nutritional status and managing evolving gastrointestinal concerns. Calorie-dense meals are recommended to support adequate growth and ensure sufficient nutrient intake. Another important aspect is pain management, as many patients report abdominal discomfort caused by flatulence, aerophagia, constipation, and gut–brain interaction disorders.37, 72 Eating disturbances may also warrant evaluation for possible laryngeal malformations. Ciachhini et al. published a case report describing the management of gastrointestinal complications in a 37-week-old boy with CFC syndrome.71 Other reports have highlighted nutritional deficiencies of key micronutrients, such as vitamin K, due to malabsorption.73
Oncology
Treatment approaches depend on the type and stage of malignancy, but data on differences in cancer management strategies in patients with RASopathies remain limited. Research on somatic mutations in cancers linked to RAS pathway activation has led to the development of targeted therapies, including MEK inhibitors (such as ulixertinib) and ERK inhibitors (such as LY3214496).74, 75 However, these agents have not yet been evaluated in animal models or patients with CFC syndrome.
Dermatology
Cardiofaciocutaneous syndrome is associated with a wide range of dermatologic symptoms, varying in severity among affected individuals. Management is primarily symptomatic and individualized, aiming to improve patient comfort and prevent complications. Typical approaches include moisturizing agents to alleviate xerosis and pruritus, topical or systemic corticosteroids for inflammatory skin lesions, keratolytic therapies for thickened or hyperkeratotic skin, and topical propranolol for the treatment of hemangiomas.76
A notable case report described a female patient with a BRAF variant who developed severe inflammatory dermatitis, characterized by erythematous lesions, yellowish palmoplantar keratoderma, and thick hyperkeratotic scalp plaques.77 The condition was refractory to conventional therapies, including corticosteroids, calcineurin inhibitors, vitamin D3 analogues, acitretin, and methotrexate. The introduction of dupilumab (300 mg every 2 weeks) resulted in marked improvement within 3 months, significantly reducing skin inflammation and hyperkeratosis. This case suggests that dupilumab may represent a potential therapeutic option for severe, treatment-resistant skin symptoms in CFC syndrome, but further research is needed to confirm its safety and effectiveness.
Urology and nephrology
Management depends on the specific condition and its severity and may range from regular monitoring and medical therapy to surgical intervention when necessary.
Ophthalmology
Early referral to a pediatric ophthalmologist is crucial, as early diagnosis and intervention significantly improve visual outcomes.78 Management strategies may include strabismus surgery, optical correction, and occlusion therapy.52
Orthopedics
Evaluation of orthopedic complications involves regular examinations by an orthopedic surgeon as well as radiographic assessment of the femoral neck–shaft angle and the lumbar and cervical regions of the spine for scoliosis. Treatment strategies may include the use of orthoses or wheelchairs, physical therapy, and regular physical activity.79
Promising therapeutic approaches
Currently, there is no causal treatment for CFC; however, 3 promising approaches emerge from experimental data:
1. MEK inhibitors, which have been used in the treatment of melanoma and non-small cell lung cancer (NSCLC). Recently, mirdametinib has been approved for use in patients with NF1.
2. Antisense oligonucleotides and A-to-I RNA editing, which may either alter the translation of mutated mRNA into a protein with altered function, thereby suppressing its activity, or modify the mutated mRNA sequence to restore the correct transcript and enable the synthesis of a protein with normal activity.
3. Prime editing, a genome-editing strategy that could represent a causal treatment targeting the underlying genetic alteration.
A substantial body of research indicates that the RAS pathway plays a key role in neurogenesis, particularly BRAF, which is involved in the proliferation, migration, and differentiation of neuronal and glial cells.80, 81, 82 This raises the possibility that early treatment – even prenatally – could potentially prevent some neurodevelopmental consequences of CFC syndrome, such as drug-resistant epilepsy (DRE), autistic-like traits, or intellectual disability. However, this hypothesis requires extensive research using cellular and mouse models specifically developed for RASopathies.
In this section, we highlight the possibility that early treatment with low doses of MEK inhibitors may prevent neurodevelopmental abnormalities and cardiac complications. However, further in vitro and in vivo studies are required to validate this approach.
In animal models, RAS/MAPK pathway inhibition in the NS mouse model prevented developmental defects (restoring normal craniofacial formation) when administered prenatally or in the early postnatal period.83, 84 However, whether these defects could be avoided if treatment were initiated later remains unclear. Given that therapy would likely need to be continued over extended periods, drugs with fewer side effects than traditional anticancer agents would represent a more suitable option. To our knowledge, no such drugs have yet been approved.85
All previously cited studies reported experimental MEK inhibitor treatment after the diagnosis of HCM or other life-threatening conditions related to RASopathies, with some patients being only a few months old. The youngest patient was 2 weeks old.86 In most cases, side effects were mild to moderate. This observation suggests that initiating MEK inhibitor therapy during the first weeks of life may not have a negative impact on subsequent development.
Off-label MEK inhibitors (especially trametinib) have shown promise in HCM associated with CFC. Low doses have been associated with improved outcomes and, in some cases, have prevented the need for surgical intervention.86, 87 Andelfinger et al. reported regression of HCM and normalization of pro-B-type natriuretic peptide levels at doses as low as 0.02–0.027 mg/kg.88 However, safety remains a concern, with reported side effects including bleeding, oliguria, diarrhea, respiratory irregularities, and mucocutaneous ulcerations.89, 90
Recent case reports highlight off-label MEK inhibitors as a promising therapeutic option for DRE in RASopathies. Selumetinib induced complete seizure cessation in a patient with NF1 despite failing to control tumor growth.91 Similarly, trametinib (0.025 mg/kg/day) led to marked seizure reduction and temporary remission in patients unresponsive to multiple antiepileptic drugs; seizures recurred after drug withdrawal due to toxicity.92, 93
Notably, 1 patient, a 6-year-old girl with CFC syndrome caused by a BRAF variant, experienced multiple seizures per day prior to treatment. However, after initiation of trametinib, electroencephalography (EEG) demonstrated a dramatic reduction in abnormal discharges 3 months after treatment. After 6 months, the patient was completely seizure-free at a dosage of 0.025 mg/kg/day.
Moreover, a study by Walsh et al. involving 59 patients with NF1 demonstrated that a 24-week course of MEK inhibitor therapy (selumetinib, trametinib, or mirdametinib) led to statistically significant improvements on psychometric assessments (BRIEF and Cogstate) compared with pre-treatment scores, particularly in domains such as visual learning, working memory, behavioral regulation, and planning.94
The off-label use of MEK inhibitors in DRE in CFC and their reported effectiveness suggest that further research is required to confirm their safety and efficacy in this condition. As of February 2025, 5 MEK inhibitors (trametinib, selumetinib, binimetinib, cobimetinib, and mirdametinib) are FDA-approved for other indications (NF1, unresectable or metastatic melanoma with BRAF V600E or V600K mutations, and NSCLC), but further research is needed to establish their safe use in CFC.89, 90 Other authors have also reported their observations regarding the use of MEK inhibitors in CFC.95 Musto et al.96 demonstrated that status epilepticus in patients with CFC may be associated with neuroinflammation; early treatment with high-dose corticosteroids resulted in neurological stabilization.
Experimental data from NF1 and NS models suggest potential pharmacological benefits of lovastatin or lamotrigine in enhancing cognitive performance, although human studies remain limited.97, 98, 99, 100
Antisense oligonucleotide therapy represents a promising future therapeutic strategy. Although no ASO-based interventions have yet been developed specifically for CFC syndrome, the success of this approach in other genetic disorders, such as spinal muscular atrophy and Duchenne muscular dystrophy,101 highlights its therapeutic potential for treating germline MAP2K2 variants. In oncology, it has been demonstrated that ASO-mediated inhibition of the long noncoding RNA MALAT1 has been shown to modulate MAPK pathway hyperactivation in melanoma.102, 103 However, translating ASO therapy to CFC will require extensive research, particularly regarding the haploinsufficiency and dosage sensitivity of the MAP2K2 gene, optimal delivery methods, toxicity profiles, allele selectivity, and the identification of an effective therapeutic window. Pharmacological treatment remains challenging due to the blood–brain barrier (BBB) limiting drug delivery, which represents a significant obstacle for potential ASO-based approaches.104
Another promising approach is prime editing, a precise genome-editing method capable of correcting base substitutions, the primary cause of most CFC cases and the underlying cause of the disease. However, this potential therapeutic approach still faces several challenges, including the creation of a suitable preclinical animal model, effective delivery to neural tissue, and the risk of immune responses to viral vectors.
Conclusions
This review summarizes all 36 published cases of CFC4, providing the most detailed overview of this ultra-rare RASopathy to date, and aims to better define its multisystem phenotype. Our analysis suggests that MAP2K2-related CFC may differ subtly from other genotypes, with PVS appearing more frequently in this condition, whereas HCM and epilepsy occur less often compared with BRAF- or MAP2K1-related disease. These observations, although limited by the small number of cases and heterogeneous clinical reporting, suggest potential genotype–phenotype correlations that warrant further investigation.
Current management remains primarily symptomatic and multidisciplinary, focusing on early detection and proactive treatment of complications. However, emerging therapies, including MEK inhibitors and experimental approaches such as ASO therapies, are beginning to reshape the treatment landscape for RASopathies. Although available data remain limited and safety concerns persist, these developments highlight the possibility of moving beyond supportive care toward targeted, disease-modifying interventions in the future.
This review emphasizes the need for consistent, detailed case reporting and international collaborative registries, which will be crucial for deepening our understanding of CFC4, clarifying genotype-specific disease patterns, and guiding the safe and effective application of novel therapies for this rare condition.
Supplementary data
The supplementary materials are available at https://doi.org/10.5281/zenodo.18876139. The package contains the following files:
Supplementary Table 1. Clinical characteristics and organ-specific manifestations of reported CFC4 cases.
Use of AI and AI-assisted technologies
Not applicable.