Abstract
This review explores the role of autoimmune mechanisms in drug-resistant epilepsy (DRE), with emphasis on etiology, mechanisms of drug resistance, and potential immunomodulatory interventions. A structured review of clinical and experimental studies was conducted to assess current knowledge on the interplay between molecular mechanisms and immune responses in DRE. Particular attention was given to the involvement of the mTOR pathway, blood–brain barrier (BBB) dysfunction, astrocyte activation, and overexpression of efflux transporters, as well as the presence of neuronal autoantibodies such as N-methyl-D-aspartate receptor (NMDAR) and GluR3. The available evidence suggests that, although conventional pharmacologic approaches fail in a subset of patients, immunologic diagnostics and targeted therapies – including intravenous immunoglobulin, corticosteroids, rituximab, and plasmapheresis – may provide clinical benefit. Seizure reduction has been reported in selected patients; however, therapeutic response remains heterogeneous and is limited by small study populations. In conclusion, early identification of autoimmune mechanisms in DRE may help optimize treatment strategies, improve patient outcomes, and reduce the long-term clinical and societal burden of the disease.
Key words: drug-resistant epilepsy, epilepsy, IVIG, autoimmune epilepsy, focal seizures
Introduction
Epilepsy is considered drug-resistant when seizures persist despite treatment with at least 2 appropriately selected, well-tolerated, and adequately dosed antiepileptic drugs (AEDs), either as monotherapy or in combination, according to the International League Against Epilepsy (ILAE) Therapeutic Working Group. A unified definition of drug-resistant focal epilepsy (DRE) is essential, as it facilitates timely diagnosis, informs treatment strategies, and supports clinical research. A clear definition also enables non-specialist clinicians to recognize and manage this condition effectively.
The ILAE framework for defining DRE is structured in 2 hierarchical levels. Level 1 categorizes response to therapy as seizure-free, treatment failure, or undetermined, with further classification based on the presence of adverse effects. This level emphasizes 2 key indicators: seizure control and treatment tolerability. Accurate assessment requires that therapies be appropriately selected, dosed, and administered, and that response be evaluated using consistent clinical criteria, such as the “rule of 3,” which defines a seizure-free period as at least 3 times longer than the longest pre-treatment seizure interval and lasting at least 12 months. Level 2 defines DRE based on evidence from level 1. Although treatment response may fluctuate over time, the likelihood of achieving remission after 2 adequate therapeutic trials is low. Therefore, failure of at least 2 appropriate medications generally defines drug-resistant epilepsy. This definition also incorporates seizure frequency and duration of observation, reflecting the persistent and dynamic nature of the disorder.
Drug-resistant epilepsy has substantial medical and social consequences. Medically, frequent seizures increase the risk of injury, cognitive decline, psychiatric disorders, adverse effects of AEDs, and sudden unexpected death in epilepsy (SUDEP). Socially, patients may experience stigma, unemployment, and increased economic burden related to treatment costs and disability. Recurrent seizures may also promote secondary epileptogenesis, further complicating disease management. Although substantial research has been conducted on DRE, autoimmune-related DRE, particularly in focal epilepsy, remains underexplored. Epilepsy affects approx. 0.5–1% of the global population, and autoimmune etiology may account for 10–20% of cases with acute onset; however, neuronal autoantibodies are identified in fewer than 10% of patients with focal DRE. Determining the precise cause of epilepsy, particularly in cases associated with hippocampal sclerosis, remains challenging. It is unclear whether hippocampal sclerosis develops idiopathically or as a consequence of autoimmune or other pathological processes. Given these challenges and gaps in the literature, this review aims to synthesize current knowledge on autoimmune-related focal DRE and provide an overview of recent research in this field.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12
Etiology of drug-resistant epilepsy
The etiology of DRE is multifactorial. In a substantial proportion of cases, apparent drug resistance reflects so-called pseudo-drug resistance, which may result from factors such as misdiagnosis of epilepsy or inappropriate drug selection. In such cases, treatment optimization may lead to clinical improvement. True drug resistance is associated with factors related to the etiopathogenesis of the epilepsy syndrome, drug-specific properties (e.g., the development of tolerance), and interindividual variability in treatment response, including genetic and environmental factors as well as patient age. Approximately 30% of patients with DRE are candidates for surgical treatment. For the remaining patients, alternative therapeutic strategies should be considered, including evaluation of the potential role of immunomodulatory therapy.
Several theories have been proposed to explain drug resistance in epilepsy. A key component of epileptogenesis involves the mechanistic target of rapamycin (mTOR) pathway, which consists of 2 protein complexes: mTORC1 and mTORC2. The mTORC1 signaling pathway serves as a central regulatory hub that integrates signals from growth factors, nutrient availability, and cellular energy status, thereby regulating cell growth and metabolism. The mTORC1 complex, composed of the mTOR kinase, the adaptor protein Raptor, mLST8, and the regulatory proteins PRAS40 and DEPTOR, is primarily activated at the lysosomal surface in response to amino acid signaling mediated by Rag GTPases and insulin-dependent signals transmitted through the PI3K–Akt–TSC1/2–Rheb axis. Once activated, mTORC1 phosphorylates key substrates, including S6 kinase 1 (S6K1) and 4E-binding protein 1 (4E-BP1), promoting translation initiation, protein synthesis, and ribosome biogenesis while inhibiting autophagy through ULK1 phosphorylation.
The mTORC2 signaling pathway serves as a critical regulator of cellular homeostasis, cytoskeletal organization, and cell survival. The mTORC2 complex, composed of mTOR kinase, Rictor, mLST8, and the regulatory proteins mSIN1 and Protor, differs from mTORC1 in both composition and activation mechanisms, being less dependent on nutrient availability and more responsive to growth factor signaling, including receptor tyrosine kinase activation. mTORC2 directly phosphorylates and activates AGC family kinases, including Akt (Ser473), SGK1, and PKC, thereby modulating cell survival, glucose metabolism, cytoskeletal reorganization, and cell polarity. Additionally, mTORC2 influences lipid homeostasis and cellular responses to oxidative stress. Under physiologic conditions, mTOR activity is negatively regulated by several factors, including the tumor suppressor proteins TSC1 (tuberous sclerosis complex 1) and TSC2 (tuberous sclerosis complex 2), which form a heterodimer. Other regulators include phosphatase and tensin homolog (PTEN) and STE20-related kinase adaptor alpha (STRADα). Mutations in genes encoding these proteins lead to dysregulation of cell proliferation, differentiation, and growth. mTORC1 regulates mRNA translation, thereby influencing synaptic function, plasticity, and the expression of neurotransmitter receptors. Consequently, the mTOR pathway contributes to both short- and long-term cellular processes, which depend on protein synthesis. Given its role in neuronal development and synaptic plasticity, hyperactivation of the mTOR pathway has been implicated in aberrant axonal growth and neurogenesis, key processes in epileptogenesis. 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29
Immune etiology of drug-resistant epilepsy
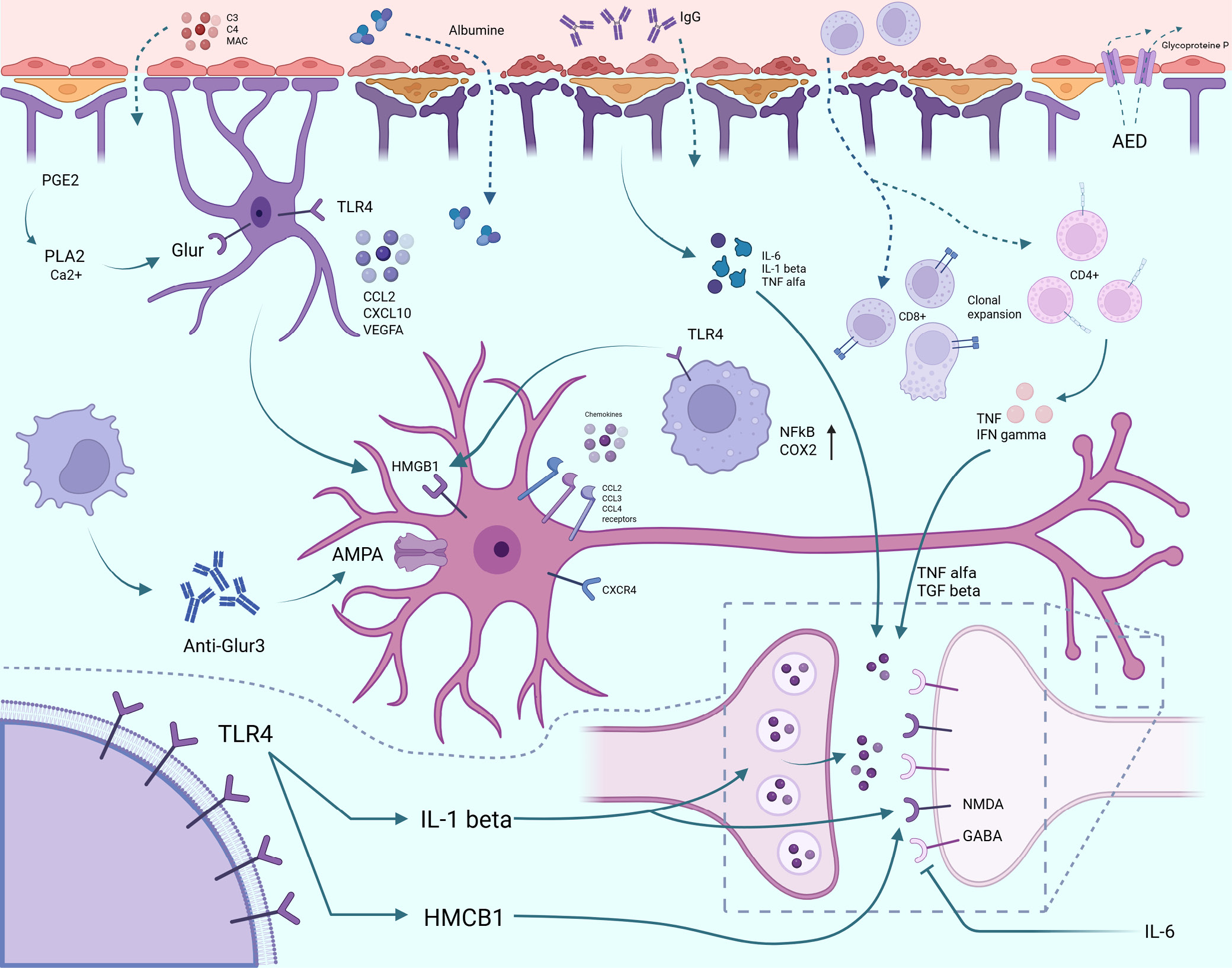
Inflammatory mediators activated during neuroinflammatory processes in DRE contribute to increased blood–brain barrier (BBB) permeability. This is associated with disruption of tight junctions and enhanced transcytosis, leading to vascular leakage within the brain. A key factor in this process is the proinflammatory phenotype of astrocytes, which express increased levels of inflammatory mediators in response to BBB dysfunction. This self-reinforcing cycle may contribute to seizure recurrence. Increased BBB permeability allows albumin to extravasate into the brain parenchyma, which may alter the pharmacokinetics and efficacy of antiseizure medications. Additionally, BBB disruption activates compensatory mechanisms, including upregulation of drug efflux transporters such as P-glycoprotein (P-gp). Increased expression of these transporters in endothelial and perivascular glial cells contributes to reduced drug penetration and pharmacoresistance.
Inflammatory processes may contribute to the etiopathogenesis of DRE, as exemplified by Rasmussen encephalitis (RE), which is characterized by drug-resistant seizures and driven by cytotoxic CD8+ T lymphocytes interacting with neurons and astrocytes. Conversely, inflammation may also arise as a consequence of epilepsy itself, as observed in temporal lobe epilepsy (TLE). Interleukin (IL)-1β appears to play a central role in the development of DRE and is produced by multiple cell types, including endothelial cells of the BBB. It may be particularly relevant during spontaneous seizures. In addition to IL-1β, increased levels of IL-6 and tumor necrosis factor alpha (TNF-α) have been reported in epileptogenic tissue. The role of chemokines and their receptors in the etiopathogenesis of DRE has also been suggested, including CCL2, CCL3, CCL4 (chemokine [C-C motif] ligands), and CXCR4 (C-X-C chemokine receptor type 4). Cyclooxygenase 2 (COX-2) also plays an important role, as its activity may promote seizure recurrence by increasing the expression of P-gp at the BBB, thereby reducing drug penetration from plasma into brain tissue and contributing to pharmacoresistance. Additionally, increased expression of toll-like receptor 4 (TLR4) and high-mobility group box 1 (HMGB1) has been observed in human neural tissue following seizures, suggesting that signaling through these molecules may contribute to seizure generation. The prevalence of drug resistance in epilepsy varies by seizure type. It is more common in patients with frequent seizures and occurs more often in focal epilepsy than in generalized epilepsy. Hippocampal sclerosis is present in 50–75% of cases of drug-resistant temporal lobe epilepsy and is associated with drug resistance in up to 89% of affected patients. A high prevalence of drug resistance is also observed in patients with temporal lobe cortical malformations (76%). When both hippocampal sclerosis and cortical malformations are present, the likelihood of drug resistance increases to 97%, whereas in other conditions it is approx. 65%.19, 30, 31, 32, 33, 34
Differences in the pathomechanisms of autoimmune drug-resistant epilepsy in children and adults
Autoimmune DRE exhibits distinct pathophysiological differences between pediatric and adult populations. In children, innate immune mechanisms appear to predominate and are characterized by sustained microglial activation and cytokine-mediated neuroinflammation. These processes often occur in the absence of identifiable autoantibodies or in the presence of antibodies of uncertain clinical significance. When detected, the most commonly reported autoantibodies include anti-N-methyl-D-aspartate receptor (anti-NMDAR), anti-myelin oligodendrocyte glycoprotein (MOG), and anti-glutamic acid decarboxylase 65 (GAD65). In pediatric patients, epileptogenesis appears to be more closely associated with neuroinflammatory mechanisms than with classical antibody-mediated autoimmunity. Inflammation in the immature, highly plastic brain may result in persistent alterations in neuronal network development and organization, thereby facilitating secondary epileptogenesis and more widespread forms of DRE.
In adults, autoimmune DRE more commonly involves adaptive immune mechanisms, with autoantibodies directed against neuronal surface antigens and ion channels, including the NMDAR, leucine-rich glioma-inactivated protein 1 (LGI1), and contactin-associated protein-like 2 (CASPR2). These antibodies directly disrupt synaptic neurotransmission and are typically associated with focal epilepsy. These age-related differences have important clinical implications, as immunotherapy may be effective in children even in the absence of defined autoantibody specificity, whereas in adults, treatment response is more closely linked to the underlying autoantibody profile.11, 35, 36, 37, 38, 39, 40
Materials and methods
This review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines and is based on available data on patients with DRE of presumed or confirmed immunologic etiology. The review also includes case reports, clinical registry data, and studies on the use of immunomodulatory therapies.
Search strategy
The literature search was conducted using the following sources: PubMed, ClinicalTrials.gov, ILAE documents, and relevant literature in neurology and neuroimmunology. Publications available up to February 2025 were included. The following English-language keywords were used: (“drug-resistant epilepsy” OR “refractory epilepsy”) AND (“autoimmune” OR “immune-mediated” OR “autoantibodies”) AND (“focal seizures” OR “temporal lobe epilepsy”) AND (“IVIG” OR “methylprednisolone” OR “immunotherapy” OR “rituximab”). Only publications with abstracts available in English or Polish were included. Additionally, data from clinical trial registries and published case reports were analyzed. The protocol was registered in PROSPERO (ID: 1086558).
Inclusion and exclusion criteria
Inclusion criteria were as follows: 1) case reports, clinical trials (ClinicalTrials.gov), meta-analyses, original studies, and systematic reviews involving patients with DRE of possible autoimmune origin; 2) studies reporting immunomodulatory treatment (intravenous immunoglobulin (IVIG), methylprednisolone, rituximab, plasmapheresis); and 3) studies including clinical assessment of treatment response. Exclusion criteria were as follows: 1) articles not available in full text; 2) studies conducted exclusively in animals; 3) studies addressing other forms of epilepsy (e.g., idiopathic generalized epilepsy); and 4) case reports not related to autoimmune mechanisms.
Data extraction and quality assessment
Data extracted from the included studies comprised patient age and sex, epilepsy type, presence of autoantibodies, type of immunomodulatory therapy, seizure frequency and severity, and clinical response. Publications were manually reviewed for eligibility according to the inclusion criteria. Study selection was performed independently by both authors (K.A.K. and E.D.), with discrepancies resolved through discussion. No formal assessment of risk of bias was performed due to the narrative nature of the review.
Synthesis methods
Data were compiled in tables: Table 137, 41, 42, 43, 44 contains case reports; Table 245, 46, 47, 48 compares clinical trials and provides a qualitative narrative analysis; Table 349, 50, 51, 52 summarizes autoimmune and inflammatory markers in focal epilepsy and DRE; and Table 453, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66 presents key findings in drug-resistant autoimmune epilepsy with focal seizures. Meta-analysis was not performed due to data heterogeneity and the limited number of comparable studies. Data and mechanisms related to the etiology of DRE are also presented graphically in Figure 1.
Data coding and analysis
Treatment response was defined as clinical improvement, including reduction in seizure frequency, improvement in electroencephalography (EEG) findings, neuroimaging results, or neurological status, according to descriptions in the original publications. Changes in EEG recordings, particularly the presence of interictal epileptiform discharges (IEDs), have been associated with the risk of seizure recurrence. A reduction in the frequency of epileptiform discharges is often considered a favorable indicator of therapeutic response; however, it does not always directly correlate with a reduction in seizure frequency. In clinical practice, EEG serves as one of several tools for assessing treatment efficacy and prognosis.67, 68 Data from studies involving patients with antineuronal antibodies (e.g., NMDAR, GluR3) were analyzed to evaluate the efficacy of IVIG, corticosteroids, rituximab, and other immunomodulatory therapies.
Only therapies with quantitative efficacy data were included in the analysis. The most frequently reported interventions were IVIG, steroids, and plasmapheresis. Disease severity and treatment effects were assessed using neurological evaluation, EEG, magnetic resonance imaging (MRI), documentation of seizure frequency before and after treatment, and clinical response as defined in each study. Variation in rating scales across studies limited the possibility of performing meta-analysis; therefore, a descriptive approach was applied. From an etiologic perspective, epilepsy may be classified as cryptogenic (of unknown origin), including cases of DRE. The occurrence of such forms and the still incompletely understood etiopathogenesis suggest that mechanisms beyond currently established concepts may contribute to disease development. These include, for example, imbalances between excitatory postsynaptic potentials (EPSPs) and inhibitory postsynaptic potentials (IPSPs), leading to excessive neuronal depolarization. One proposed hypothesis is that autoimmune processes may underlie the development of certain forms of epilepsy. Early studies focused on alterations in the levels and ratios of nonspecific immunoglobulins, such as immunoglobulin G (IgG) and immunoglobulin M (IgM); however, these changes are now considered more likely to be consequences of seizures rather than their primary cause.45, 69 Rasmussen encephalitis is a prototypical example of an autoimmune disease associated with focal seizures. In this condition, autoantibodies against the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, particularly the GluR3 subunit, have been described. These autoantibodies may also contribute to complement activation, including components such as C3, C8, and the membrane attack complex (MAC). Under physiologic conditions, IgG antibodies do not cross the BBB; however, disruption of BBB integrity may allow anti-GluR3 antibodies to access neuronal targets and contribute to disease pathogenesis. Recent evidence also highlights the role of cellular immune responses, with increased infiltration of CD8+ T lymphocytes. CD4+ T cells may also undergo clonal expansion and secrete proinflammatory cytokines, including tumor necrosis factor (TNF) and interferon gamma (IFN-γ).45, 70, 71, 72
Some autoimmune diseases are also characterized by an increased frequency of seizures. This is the case with systemic lupus erythematosus (SLE), where there is an increase in the level of antiphospholipid antibodies (aPL), antibodies against cardiolipin (aCL), B2-glycoprotein, dsDNA, and antinuclear antibodies (ANA). The increase in these antibodies correlates with the frequency of seizures in patients with SLE, which is estimated to be 28%, while the occurrence of seizures ranges from 17% to 37%.73, 74
Other conditions potentially associated with autoimmune epileptogenesis include:
− Stiff-person syndrome, characterized by elevated levels of autoantibodies against glutamic acid decarboxylase (GAD), the enzyme responsible for γ-aminobutyric acid (GABA) synthesis;
− West syndrome and Lennox–Gastaut syndrome, in which the etiology remains incompletely understood; however, a more favorable response to immunotherapy than to standard antiseizure medications suggests a potential immune-mediated component;
− Landau–Kleffner syndrome (LKS), in which clinical improvement has been observed following treatment with IVIG, along with evidence of intrathecal IgG synthesis, as indicated by elevated cerebrospinal fluid (CSF) IgG levels;
− Hashimoto encephalopathy;
− Channelopathies.
Some antiseizure medications have immunosuppressive effects, particularly carbamazepine, phenytoin, and valproate. These agents may reduce lymphocyte counts, alter the CD4+/CD8+ ratio, and decrease immunoglobulin levels, including IgG, immunoglobulin A (IgA), and IgM. Their effects on cytokine levels remain less well defined. Evidence supporting the efficacy of IVIG therapy has been reported. In a study by Biliau et al.,58 involving 13 children with DRE, a significant reduction in seizure frequency was observed in 4 patients, a moderate reduction (20% to 50%) in 3 patients, no change in 5 patients, and an increase in seizure frequency in 1 patient. Intravenous immunoglobulig therapy has also been investigated in RE. Hart et al.74 evaluated corticosteroid and IVIG therapy in 9 patients with RE, of whom 8 who received IVIG demonstrated a reduction in seizure frequency.63, 75
The impact of epilepsy on oral health, clinical observations, and implications
Epilepsy in adults is associated with impaired oral health. This is primarily related to difficulties in motor coordination and reduced manual dexterity, which may hinder effective oral hygiene practices. Studies have shown that individuals with epilepsy, compared with healthy controls, exhibit poorer oral health and a higher prevalence of gingival overgrowth. These patients more frequently require dental interventions, including root canal treatment, dental fillings, and tooth extractions. A similar pattern is observed in pediatric populations. Children with epilepsy are more prone to gingivitis, anterior tooth injuries, and increased dental plaque accumulation on permanent teeth compared with their healthy peers.
Children with epilepsy have been reported to exhibit reduced relative abundance of Bacillota and Bacteroidota, along with an increased presence of Actinomycetota in the oral microbiome compared with healthy controls. One contributing factor to poorer oral health in this population is the reduced frequency of dental visits, often related to increased anxiety about dental procedures. Additional factors include impaired motor function affecting oral hygiene, lower socioeconomic status, and oral injuries sustained during seizures, such as mucosal and tongue biting or dental trauma. Seizure-related injuries may also involve the temporomandibular joint (TMJ). Seizures may manifest with involuntary movements affecting this joint, such as oral automatisms and masticatory movements, particularly in focal seizures originating in the frontal or temporal lobes. Such oromandibular automatisms may result in TMJ dislocation. Case reports have described TMJ dislocation occurring during seizure episodes, including in a female patient. Additionally, involuntary facial movements may occur as adverse effects of certain antiseizure medications, manifesting as drug-induced dyskinesias. For example, 1 case report described involuntary facial and limb movements following levetiracetam administration, without corresponding epileptiform discharges on EEG.
Antiseizure medications (ASMs) may exert various effects on oral health. Studies suggest that newer ASMs – such as oxcarbazepine, levetiracetam, ethosuximide, clobazam, lamotrigine, zonisamide, perampanel, topiramate, vigabatrin, and lacosamide – may be associated with slight improvements in oral health indices, including decayed, missing, and filled surfaces (DMFS), plaque index (PI), and decayed, missing, and filled teeth (DMFT), compared with regimens involving older ASMs alone (e.g., phenytoin (PHT), valproate, carbamazepine, and phenobarbital). Notably, a reduced incidence of gingivitis and periodontitis has been observed in patients treated with newer ASMs. Gingival hyperplasia, in particular, is a well-established complication of chronic PHT therapy. The oral cavity may play an important role in the development of psychiatric and neuropsychiatric disorders, including epilepsy. One of the key mechanisms underlying this association involves the oral microbiota. Through the oral microbiota–brain axis and the gut–brain axis, oral microorganisms may influence neurotransmission both directly and indirectly. The microbiota may also access the central nervous system via neural pathways, including the trigeminal and olfactory nerves. Oral diseases may disrupt the mucosal barrier, facilitating the translocation of bacteria and endotoxins into the bloodstream and across the BBB, potentially triggering neuroinflammatory processes associated with epilepsy.
Patients with epilepsy have been reported to exhibit increased oral microbial diversity compared with healthy controls, characterized by higher α-diversity and significant differences in β-diversity between groups. Specifically, reduced abundance of 14 bacterial genera has been observed, including Schaalia, Neisseria, and Peptostreptococcus, along with increased abundance of 26 genera, such as Kluyvera, Granulicatella, and Streptococcus. Importantly, no significant differences in oral microbiota composition have been observed before and after seizure control. Increased abundance of Streptococcus has also been reported in the gut microbiota of patients with epilepsy and correlates with elevated levels of IL-6 and TNF-α, cytokines involved in neuroinflammatory processes. Interleukin-6 and TNF-α are well-established salivary biomarkers, e.g., in the diagnosis of oral and oropharyngeal squamous cell carcinoma, and their elevated levels may also indicate odontogenic infections in the maxillofacial region. Beyond these contexts, increased concentrations of these cytokines may reflect ongoing neuroinflammation. Although not currently used as standard biomarkers for epilepsy in clinical practice, growing evidence suggests their potential utility in monitoring inflammatory states in patients with epilepsy.76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91
Autoimmune and paraneoplastic antibody-associated encephalitides and blood biomarkers in epilepsy
Autoimmune encephalitis (AE) comprises a heterogeneous group of disorders in which immune responses directed against neuronal antigens result in central nervous system inflammation and a broad spectrum of neurologic and psychiatric manifestations. Target antigens may include neuronal surface or synaptic proteins (e.g., the NMDAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), or components of the voltage-gated potassium channel complex, such as leucine-rich glioma-inactivated protein 1 (LGI1) and CASPR2) as well as intracellular antigens. Antigen localization has important implications for pathophysiology, clinical presentation, and prognosis.
Among these, antibodies against the NMDAR are among the best characterized. They are frequently associated with psychiatric symptoms, movement disorders, autonomic instability, and seizures, with multiple clinical series demonstrating a high prevalence of seizures in this condition. Less common but clinically relevant are antibodies targeting the AMPAR, which have been reported in cases of limbic encephalitis (LE), often accompanied by seizures, although the precise incidence varies across studies. Within the voltage-gated potassium channel (VGKC) complex, antibodies against LGI1 and CASPR2 are associated with distinct clinical phenotypes. LGI1 antibody-associated AE is strongly linked to faciobrachial dystonic seizures (FBDS) – brief dystonic movements involving the face and arm – that often precede LE. In contrast, CASPR2 antibody-associated AE exhibits more heterogeneous neurologic involvement, sometimes including both central and peripheral features. Seizures occur less frequently than in LGI1-associated AE but remain a recognized component of the disease. Antibodies directed against dipeptidyl peptidase-like protein 6 (DPPX) are rare but clinically important causes of AE characterized by neuronal hyperexcitability. Case reports and series indicate that DPPX autoimmunity may present with myoclonus, startle phenomena, gastrointestinal symptoms, and autonomic instability. Seizures have been reported in up to 30–50% of cases, reflecting increased neuronal excitability, particularly within the hippocampus.
Diagnosis of AE relies on an integrated approach combining clinical assessment, MRI, EEG, CSF analysis, and detection of specific antibodies in serum and/or CSF. Although antibody detection supports the diagnosis, results must be interpreted in the appropriate clinical context, as isolated seropositivity may occur without overt autoimmune inflammation. Consensus diagnostic algorithms emphasize stepwise evaluation and timely initiation of immunotherapy. Recent studies highlight the utility of fluid biomarkers of central nervous system injury, including neurofilament light chain (NfL) and glial fibrillary acidic protein (GFAP). Elevated levels of NfL in serum and CSF reflect axonal damage and correlate with disease activity and severity, whereas GFAP serves as a marker of astrogliosis and glial activation. These biomarkers may complement immunologic and neuroimaging findings, providing insight into the extent of neuronal injury and potential response to therapy; however, they are not yet diagnostic substitutes for antibody testing.
In addition to classic AE, paraneoplastic neurologic syndromes (PNS) mediated by onconeural antibodies should be considered. Antibodies such as anti-Hu (ANNA-1), anti-Yo, anti-Ma2, and anti-collapsin response mediator protein 5 (CRMP5; CV2) target intracellular antigens and are associated with cytotoxic T cell-mediated neuronal injury. Their detection should prompt an urgent search for an underlying malignancy, as PNS often precede tumor diagnosis. Current guidelines recommend combining antibody testing with whole-body positron emission tomography/computed tomography (PET/CT) for tumor detection and staging.
Autoimmune and paraneoplastic encephalitides constitute an important and often reversible cause of seizures and encephalopathy. Accurate diagnosis requires integration of clinical, electrophysiologic, imaging, and immunologic data. Novel biomarkers, such as NfL and GFAP, together with traditional paraneoplastic antibody panels, enhance diagnostic precision and prognostic assessment. Early recognition and initiation of immunotherapy or tumor-directed treatment significantly improve outcomes, underscoring the need for clinician awareness and prompt diagnostic evaluation.61, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109 In addition to human studies, animal models of AE provide complementary evidence for the pathogenic role of neuronal autoantibodies. Passive transfer and active immunization models targeting antigens such as the NMDAR, LGI1, and CASPR2 replicate key electrophysiologic and behavioral features observed in patients, including increased seizure susceptibility and background EEG alterations. These findings highlight the translational relevance of preclinical models for understanding antibody-mediated mechanisms in DRE.
Limitations of the study
This review has several limitations that should be considered when interpreting its findings. First, the available studies on autoimmune DRE with focal seizures are limited and often involve small sample sizes, restricting the generalizability of the results. Second, heterogeneity in study design, patient populations, diagnostic criteria, and treatment protocols precluded quantitative synthesis or meta-analysis, necessitating a primarily descriptive approach. Third, many included studies are case reports or observational studies, which are susceptible to reporting bias and lack randomization. Fourth, standardized outcome measures for seizure reduction and response to immunotherapy are often lacking, making comparisons across studies challenging. These limitations underscore the need for larger, well-designed clinical trials and standardized protocols to better assess the role of autoimmune mechanisms and immunotherapies in DRE.
Conclusions
This review demonstrates that autoimmune mechanisms represent a clinically relevant and potentially modifiable contributor to DRE in a distinct subset of patients. Evidence summarized in this article indicates that neuroinflammation, BBB dysfunction, mechanistic mTOR pathway hyperactivation, and neuronal autoantibodies – particularly against the NMDAR and GluR3 – play a significant role in both epileptogenesis and resistance to antiseizure medications. Importantly, clinical data suggest that immunomodulatory therapies, including intravenous immunoglobulin, corticosteroids, rituximab, and plasmapheresis, can lead to meaningful seizure reduction in selected patients, particularly those with identifiable autoimmune markers or inflammatory features.
The main conclusion of this review is that DRE should not be regarded as a purely pharmacologic failure but rather as a heterogeneous condition in which immune-mediated mechanisms may directly contribute to seizure persistence in a subset of cases. Early recognition of an autoimmune background – through targeted antibody testing, inflammatory biomarkers, and clinical phenotyping – may enable timely initiation of immunotherapy and improve seizure control, neurologic outcomes, and quality of life. However, the available evidence remains limited by small cohort sizes, heterogeneous study designs, and a lack of standardized treatment protocols. Therefore, although immunotherapy cannot be universally recommended for all patients with DRE, it should be actively considered in those with focal seizures and features suggestive of an autoimmune etiology. Future prospective studies and randomized controlled trials (RCTs) are needed to establish clear diagnostic criteria, identify reliable predictive biomarkers, and optimize treatment strategies for autoimmune-associated DRE.
Use of AI and AI-assisted technologies
ChatGPT was used in editing and translating sections of the article.