















Abstract
Background. The precise causal relationship between alterations in the gut microbiota, microbiota-derived metabolites, and the development of diffuse large B-cell lymphoma (DLBCL) remains unclear.
Objectives. To investigate the potential causal relationships between gut microbiota, microbiota-derived metabolites, and DLBCL.
Materials and methods. Genetic data on gut microbiota were obtained from the MiBioGen consortium, while data on microbiota-derived metabolites were sourced from the TwinsUK and KORA studies. Summary statistics for DLBCL were retrieved from FinnGen. Mendelian randomization (MR) analysis was performed, with inverse-variance weighting (IVW) used as the primary analytical method. Sensitivity analyses included Cochran’s Q test, the MR-Egger intercept test, and MR-PRESSO. Reverse MR analysis was conducted to assess potential bidirectional causal relationships between gut microbiota and DLBCL. Bayesian weighted MR (BWMR) was applied for additional validation to enhance the robustness of the findings.
Results. Among 196 gut microbial taxa analyzed, Bilophila (odds ratio (OR) = 1.777, 95% confidence interval (95% CI): 1.053–3.000, p = 0.031) was associated with an increased risk of DLBCL. In contrast, Alistipes (OR = 0.521, 95% CI: 0.311–0.873, p = 0.013) and Ruminococcaceae UCG011 (OR = 0.749, 95% CI: 0.574–0.978, p = 0.034) were associated with a reduced risk. Reverse MR analysis demonstrated a positive association between DLBCL risk and the abundance of Anaerofilum (OR = 1.087, 95% CI: 1.008–1.173, p = 0.031). Negative associations were observed between DLBCL risk and the abundance of Deltaproteobacteria (OR = 0.959, 95% CI: 0.922–0.997, p = 0.037), Desulfovibrionales (OR = 0.959, 95% CI: 0.922–0.998, p = 0.041), Oxalobacteraceae (OR = 0.914, 95% CI: 0.843–0.992, p = 0.031), and Oxalobacter (OR = 0.909, 95% CI: 0.837–0.988, p = 0.024). Analysis of microbiota-derived metabolites identified a causal association between indolepropionate (OR = 0.296, 95% CI: 0.131–0.669, p = 0.003) and reduced DLBCL risk, whereas 7-alpha-hydroxy-3-oxo-4-cholestenoate (7-HOCA) (OR = 9.561, 95% CI: 1.426–64.088, p = 0.020) was associated with an increased risk. No evidence of directional pleiotropy or heterogeneity was detected.
Conclusions. This MR study provides evidence that specific gut microbial taxa and microbiota-derived metabolites may causally influence the risk of DLBCL.
Key words: metabolomics, gut microbiota, diffuse large B-cell lymphoma, Mendelian randomization, Bayesian statistics
Background
Non-Hodgkin lymphoma (NHL) comprises a heterogeneous group of malignant lymphoproliferative disorders arising from lymphocytes.1 In recent decades, the incidence of NHL has increased significantly, and it now ranks as the 11th most common malignancy worldwide, contributing substantially to the global cancer burden.1, 2 Diffuse large B-cell lymphoma (DLBCL) is the most prevalent subtype of NHL, accounting for approx. 30–58% of cases, with notable geographic variation in its distribution.3, 4
Although DLBCL is considered clinically aggressive, standard treatment regimens generally yield favorable outcomes, with 60–70% of patients achieving a 5-year overall survival (OS) rate. Nevertheless, approx. 30–40% of patients eventually experience relapse or treatment failure, partly due to an incomplete understanding of the disease’s underlying pathogenic mechanisms.3
Although the etiology of DLBCL remains incompletely elucidated, accumulating evidence suggests its development involves complex interactions between genetic susceptibility, immune dysregulation, and environmental exposures.5 In this context, the proposed microbiota–gut–lymphoma axis highlights the potential mechanistic link between microbial alterations and lymphomagenesis.6, 7 The human gastrointestinal tract contains approx. 100 trillion microorganisms,8, 9 which contribute to host physiology by enhancing nutrient absorption,10 providing pathogen defense,11 maintaining intestinal barrier function, modulating epithelial differentiation,12 and regulating immune responses.13 However, disruption of these microbial communities, referred to as dysbiosis, may impair normal physiological functions.9 Characterized by a reduction in beneficial microbes and/or an increase in pathogenic species, dysbiosis has been linked to chronic inflammation, immune dysregulation, and tumor development.7, 14 Emerging evidence indicates that the gut microbiota contributes not only to the development of gastrointestinal malignancies but also to oncogenesis at extraintestinal sites.14, 15, 16, 17, 18, 19 Recent studies have identified distinct gut microbial profiles in newly diagnosed DLBCL patients compared with healthy controls,18, 20, 21, 22 with specific alterations in microbial composition correlating with clinical outcomes.22, 23 Furthermore, accumulating evidence indicates that microbial metabolites may influence DLBCL progression by modulating immunometabolic pathways.24
Although current research has demonstrated correlations between gut microbial profiles, microbial metabolites, and DLBCL, causal relationships remain unproven, as most existing studies have employed case–control designs and therefore cannot establish temporal relationships between microbial alterations and lymphomagenesis.22 In addition, observational studies examining gut microbiota–DLBCL associations are subject to inherent limitations due to multiple confounding factors, including age, environmental exposures, diet, lifestyle, comorbidities, and therapeutic interventions, which may substantially bias association estimates.25 While randomized controlled trials represent the gold standard for causal inference, their implementation is often limited by ethical and practical constraints. Therefore, Mendelian randomization (MR) and other causal inference methodologies may offer alternative approaches to elucidate potential microbiome–lymphoma relationships. Mendelian randomization is an epidemiological method that strengthens causal inference by using genetic variants as instrumental variables (IVs), leveraging the random allocation of alleles at conception to minimize confounding from postnatal environmental factors.26 This approach has gained increasing utility in investigating potential causal links between gut microbiota composition and various cancers.27, 28
This study employed a two-sample MR framework to systematically investigate potential causal relationships between gut microbiota composition and DLBCL risk. We aimed to elucidate the role of specific gut microbial taxa in DLBCL pathogenesis while simultaneously assessing the bidirectional nature of microbiota–lymphoma associations. Furthermore, we evaluated potential causal relationships between gut microbiota–derived metabolites and DLBCL development. By applying robust MR methodologies, our findings may provide novel insights into gut microbiome–DLBCL interactions and potentially inform the development of microbiota-targeted strategies for DLBCL prevention and clinical management.
Objectives
This study employed MR to systematically investigate causal relationships between gut microbiota composition and DLBCL risk, with a particular focus on identifying potentially pathogenic microbial taxa and their contributions to DLBCL lymphomagenesis. Our findings may provide insights into the development of microbiome-modulating interventions for DLBCL prevention and treatment.
Materials and methods
Study design
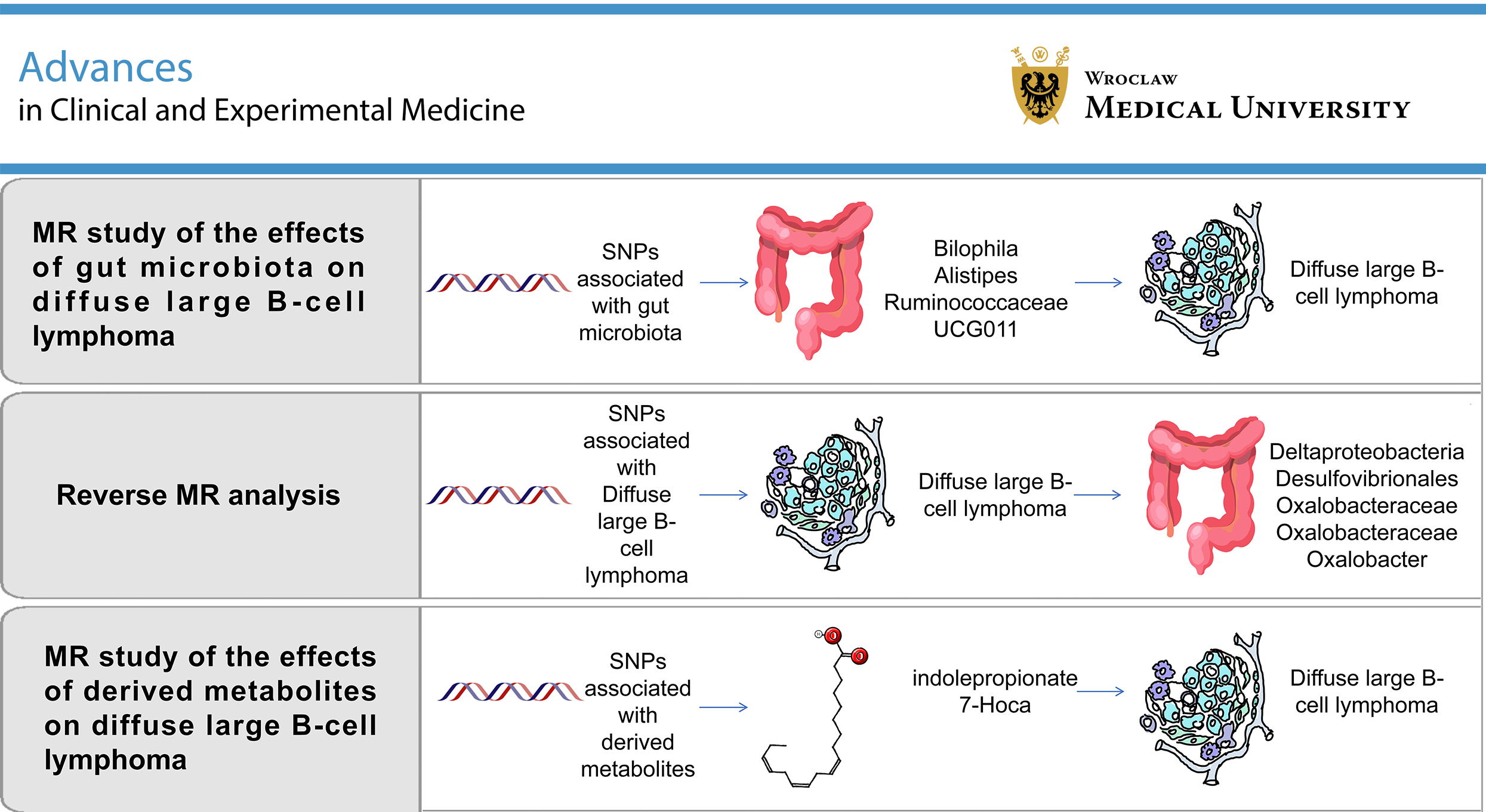
This MR study was conducted in accordance with the Epidemiology-MR (STROBE-MR) guidelines. We applied both conventional two-sample MR and Bayesian weighted MR (BWMR) to assess causal relationships between gut microbiota composition, microbial metabolites, and DLBCL risk. A schematic overview of the study design is presented in Figure 1. Instrumental variables were selected as single nucleotide polymorphisms (SNPs) significantly associated with the exposures from genome-wide association study (GWAS) datasets. Mendelian randomization analyses were performed using the TwoSampleMR package in the R statistical environment (R Foundation for Statistical Computing, Vienna, Austria). Comprehensive sensitivity analyses, including heterogeneity tests, pleiotropy assessments, and leave-one-out analyses, were conducted to evaluate the robustness of the findings. In addition, BWMR provided further validation of the causal estimates through its robust Bayesian framework. Reverse MR analyses were also performed to evaluate potential reverse causality between gut microbiota and DLBCL. Our analytical framework satisfied the 3 core assumptions of MR:
1. The SNPs used as IVs must be robustly associated with the exposure;
2. The IVs must be independent of confounders influencing both the exposure and the outcome;
3. The IVs must affect the outcome exclusively through the exposure (i.e., absence of horizontal pleiotropy).
Data sources
The gut microbiota dataset was obtained from the MiBioGen consortium GWAS (https://mibiogen.gcc.rug.nl), comprising 18,340 individuals of predominantly European ancestry across 24 cohorts.29 This resource provided 16S rRNA gene sequencing profiles paired with genome-wide genotyping data, identifying 211 bacterial taxa across 5 taxonomic levels: 9 phyla, 16 classes, 20 orders, 35 families, and 131 genera. To enhance analytical robustness, we excluded 3 taxonomically undefined families and 12 unclassified genera that could introduce annotation inconsistencies or noise into SNP-exposure associations. Metabolite GWAS data were obtained from 2 European population-based studies (TwinsUK and KORA), comprising 7,824 participants with quantitative profiles of 486 circulating metabolites. From this dataset, we systematically identified and extracted 77 gut microbiota-derived metabolites through annotation based on the Human Metabolome Database (https://hmdb.ca).
The DLBCL summary statistics were obtained from the latest release (R10; published December 18, 2023) of the FinnGen consortium, specifically from the “Diffuse large B-cell lymphoma” dataset, comprising 1,050 cases and 314,193 controls. As this study exclusively utilized de-identified, publicly available data from established consortia, no additional institutional ethics approval was required. When treating gut microbiota and their derived metabolites as exposures and DLBCL as the outcome, the selection of IVs was based on the following criteria: 1) Based on prior literature, we set the significance threshold at p < 1.0 × 10–5 to maximize the number of genetic instruments for exposures30; 2) To ensure independence between IVs, LD pruning was conducted using a threshold of r2 < 0.001 and a clumping window of 10,000 kb30; 3) Exposure and outcome datasets were harmonized, with palindromic SNPs removed. Single nucleotide polymorphisms with F-statistics ≤10 were excluded to ensure instrument strength, as the F-statistic reflects the strength of the association between IVs and the exposure.31 For the reverse MR, in which DLBCL was treated as the exposure and gut microbiota as the outcome, the significance threshold was set at p < 5 × 10–6. All other IV selection criteria were consistent with those applied in the forward MR analysis.
Statistical analyses
A range of MR methodologies was applied to investigate the potential causal association between gut microbiota and DLBCL. For bacterial genera with only a single available IV, the Wald ratio method was used to estimate causal effects.32 When multiple IVs were available, 5 complementary MR methods were applied: inverse-variance weighted (IVW), MR-Egger regression, weighted median, simple mode, and weighted mode. Under the assumption of no horizontal pleiotropy, the IVW method provides the most precise estimates and was therefore selected as the primary analytical approach in this study.33 MR-Egger regression, which assumes that instrument strength is independent of direct effects (InSIDE assumption), was used as a sensitivity analysis to assess horizontal pleiotropy.34 The weighted median method can provide consistent causal estimates even when up to 50% of the genetic instruments are invalid.35 A key excerpt of the R script used for the data analysis is provided in the shared raw data. The F-statistic for each SNP was calculated using the formula F = β2/SE2, and SNPs with F-statistics ≤10 were excluded to minimize weak instrument bias. To assess the robustness of MR estimates, several sensitivity analyses were subsequently performed. Cochran’s Q test was used to detect heterogeneity among IVs, with p < 0.05 indicating statistical significance.36 Horizontal pleiotropy was evaluated using both the MR-Egger intercept test (estimating the average pleiotropic effect) and the MR-PRESSO global test (detecting outlier SNPs).34, 37 In addition, a leave-one-out analysis was conducted to determine whether any single IV had a disproportionate influence on causal effect estimates.38 The BWMR was employed to account for uncertainties arising from polygenicity and to address violations of IV assumptions by controlling for outliers.39
Results
MR results
The IVW method was used as the primary analytical approach, with BWMR providing complementary validation. The IVW analysis identified 4 gut microbial taxa showing statistically significant associations with DLBCL risk (p < 0.05). Specifically, Bilophila (odds ratio (OR) = 1.777, 95% confidence interval (95% CI): 1.053–3.000, p = 0.031) and Desulfovibrionaceae (OR = 1.577, 95% CI: 1.003–2.487, p = 0.049) were positively associated with an increased risk of DLBCL. In contrast, Alistipes (OR = 0.521, 95% CI: 0.311–0.873, p = 0.013) and Ruminococcaceae UCG011 (OR = 0.749, 95% CI: 0.574–0.978, p = 0.034) were inversely associated with DLBCL risk (Table 1). Scatter plots illustrating the nominally significant associations (p < 0.05) identified through IVW analysis are presented in Figure 2A–C.
The BWMR validation confirmed 3 of these associations and refined their effect estimates. Alistipes (OR = 0.573, 95% CI: 0.341–0.964; p = 0.036) and Ruminococcaceae UCG011 (OR = 0.747, 95% CI: 0.565–0.987; p = 0.040) maintained their inverse associations with DLBCL risk, whereas Bilophila demonstrated a stronger positive association (OR = 2.005, 95% CI: 1.238–3.248; p = 0.005). In contrast, the association for Desulfovibrionaceae was no longer statistically significant in the BWMR analysis (OR = 1.342, 95% CI: 0.846–2.130; p = 0.212), and was therefore not considered in subsequent analyses (Table 1).
Moreover, our MR analysis of gut microbiota–derived metabolites identified 3 significant associations with DLBCL risk. Two metabolites, 3-(4-hydroxyphenyl)lactate (OR = 0.174, 95% CI: 0.030–0.784; p = 0.023) and indolepropionate (OR = 0.296, 95% CI: 0.131–0.669; p = 0.003), were inversely associated with DLBCL risk, suggesting potential protective effects. In contrast, 7α-hydroxy-3-oxo-4-cholestenoate (7-HOCA) (OR = 9.561, 95% CI: 1.426–64.088; p = 0.020) exhibited a strong positive association with DLBCL risk, indicating a potential risk-promoting role (Table 2). BWMR validation confirmed 2 of these associations. Indolepropionate (OR = 0.256, 95% CI: 0.100–0.650; p = 0.004) maintained its inverse association with DLBCL risk, whereas 7-HOCA (OR = 10.577, 95% CI: 1.275–87.729; p = 0.029) demonstrated an even stronger positive association. However, 3-(4-hydroxyphenyl)lactate (OR = 0.230, 95% CI: 0.040–1.314; p = 0.098) did not retain statistical significance in the BWMR analysis and was therefore not considered in further interpretation (Table 2). Scatter plots depicting the significant metabolite–DLBCL associations identified through BWMR analysis are presented in Figure 2D–E. Forest plots visually summarize all significant microbiota–DLBCL causal relationships (Supplementary Fig. 1). Funnel plots demonstrated symmetrical distributions of causal estimates, indicating minimal evidence of directional pleiotropy that could bias the results (Supplementary Fig. 2).
Sensitivity analysis
Sensitivity analyses consistently supported the robustness of our findings. Cochran’s Q test revealed no significant heterogeneity among the selected IVs (Table 1, Table 2). Additionally, the MR-Egger intercept test provided no evidence of significant horizontal pleiotropy. The MR-PRESSO global test (p > 0.05) identified no outlier variants among the gut microbial taxa, microbiota-derived metabolites, or DLBCL associations, further supporting the absence of horizontal pleiotropy (Table 1, Table 2). Finally, the leave-one-out analysis confirmed the stability of the estimates, demonstrating that no individual variant exerted a disproportionate influence on the causal associations (Supplementary Fig. 3).
Reverse MR results
In the reverse MR analysis evaluating DLBCL as exposure and gut microbiota as outcome, we initially identified 12 IVs, excluding those with weak instrument effects. Using IVW as the primary statistical method, DLBCL was found to be associated with a higher abundance of Anaerofilum (OR = 1.087, 95% CI: 1.008–1.173, p = 0.031) and a lower abundance of Deltaproteobacteria (OR = 0.959, 95% CI: 0.922–0.997, p = 0.037), Desulfovibrionales (OR = 0.959, 95% CI: 0.922–0.998, p = 0.041), Desulfovibrionaceae (OR = 0.960, 95% CI: 0.923–0.999, p = 0.045), Oxalobacteraceae (OR = 0.914, 95% CI: 0.843–0.992, p = 0.031), and Oxalobacter (OR = 0.909, 95% CI: 0.837–0.988, p = 0.024) (Table 3). The BWMR analysis provided further validation of the causal relationships between DLBCL and specific gut microbial taxa. This robust approach confirmed significant associations of DLBCL with Deltaproteobacteria (OR = 0.958, 95% CI: 0.920–0.999, p = 0.042), Desulfovibrionales (OR = 0.959, 95% CI: 0.920–0.999, p = 0.046), Oxalobacteraceae (OR = 0.911, 95% CI: 0.838–0.990, p = 0.027), Anaerofilum (OR = 1.091, 95% CI: 1.008–1.181, p = 0.030), and Oxalobacter (OR = 0.905, 95% CI: 0.832–0.986, p = 0.022). However, the association with Desulfovibrionaceae (OR = 0.960, 95% CI: 0.921–1.001, p = 0.051) narrowly missed the statistical significance threshold and was consequently excluded from final interpretation. Scatter plots of SNP effects supported the causal associations between DLBCL risk and gut microbiota abundance, as shown in Supplementary Fig. 4.
Forest plots demonstrated significant effects of DLBCL on specific gut microbial taxa in the reverse MR analysis (Supplementary Fig. 5). Funnel plots derived from the reverse MR analysis demonstrated a symmetrical distribution of variant effects, suggesting a low likelihood of directional pleiotropy (Supplementary Fig. 6). Importantly, sensitivity analyses performed for the reverse MR findings provided no evidence of significant heterogeneity or horizontal pleiotropy, with all relevant statistics summarized in Table 3. The leave-one-out analysis further confirmed the stability of the estimates, indicating that no single IV exerted a disproportionate influence on the causal effect estimates (Supplementary Fig. 7).
Discussion
In this study, we applied MR to systematically investigate the causal relationships between gut microbial features, microbiota-derived metabolites, and DLBCL risk. The findings identified 3 bacterial genera – Alistipes, Ruminococcaceae UCG011, and Bilophila – along with 2 microbiota-derived metabolites, indolepropionate and 7-HOCA, that demonstrated significant causal associations with DLBCL risk. Furthermore, reverse MR analysis suggested that DLBCL may reciprocally influence gut microbial composition, with putative causal effects observed for 1 bacterial class (Deltaproteobacteria), 1 order (Desulfovibrionales), 1 family (Oxalobacteraceae), and 2 genera (Anaerofilum and Oxalobacter). The gut microbiota, comprising more than 1,000 taxonomic units residing in the intestinal tract, is increasingly recognized as a vital “virtual organ” due to its structural complexity and partially heritable characteristics. This microbial community plays a pivotal role in maintaining host physiological homeostasis through diverse direct and indirect mechanisms.40 Growing evidence suggests that perturbations in gut microbial ecology contribute to the pathogenesis of various diseases, including hematological malignancies.41, 42 Recent studies have further implicated gut microbiota dysbiosis in lymphomagenesis and disease progression.43, 44 Supporting this connection, Lin et al. reported distinct gut microbiota profiles in untreated DLBCL patients, characterized by significantly elevated abundances of Proteobacteria, Escherichia–Shigella, Roseburia, and Alistipes compared with healthy controls.18 In contrast, Li et al. observed a significant depletion of Roseburia in DLBCL patients.20 Further clinical observations indicated that patients experiencing treatment-related adverse events exhibited higher levels of Enterobacteriaceae, whereas those without adverse events showed relative enrichment of Prevotellaceae and Oscillospiraceae.22 Moreover, a significant difference in Enterobacteriaceae abundance was observed between patients with disease relapse or progression and those in remission.22 Recent investigations have identified significant microbial shifts in treatment-naïve patients, characterized by increased levels of Bacteroidetes and decreased levels of Firmicutes compared with healthy controls.43 Firmicutes, particularly members of the families Ruminococcaceae and Lachnospiraceae, are major butyrate producers in the human colon.45, 46 In our study, Ruminococcaceae UCG011, a butyrate-producing bacterial genus, was inversely associated with DLBCL risk (OR = 0.749). This protective association suggests potential anti-lymphoma effects that may operate through multiple butyrate-mediated mechanisms. As the primary short-chain fatty acid (SCFA) generated through microbial fermentation of dietary fiber, butyrate serves as a key energy source for colonocytes while also modulating immune responses and enhancing intestinal barrier integrity.47 Moreover, Wei et al. demonstrated that butyrate exerts potent anti-tumor effects by functioning as a histone deacetylase (HDAC) inhibitor, thereby promoting histone acetylation and inducing apoptosis of malignant cells through epigenetic modulation of gene expression pathways.24 The anti-tumor effects of butyrate may be particularly relevant in DLBCL, given the characteristic overexpression of HDAC isoforms observed in this malignancy.48, 49 Similar HDAC-mediated oncogenic processes have been reported across multiple malignancies, in which butyrate’s HDAC-inhibitory activity promotes tumor-suppressive histone acetylation and apoptosis.50, 51
Existing evidence also indicates that butyrate regulates key oncogenic pathways including mitochondrial and extrinsic apoptosis, G protein-coupled receptor (GPR41/43/109a) signaling, Wnt signaling, and protein kinase C pathway.52 Particularly relevant to lymphomagenesis, Lu et al. reported that butyrate-producing Eubacterium rectale suppresses tumor necrosis factor (TNF) production and subsequent TLR4/MyD88/NF-κB activation in B cells, which may contribute to reduced lymphoma incidence.23 Our current analysis did not detect significant associations of E. rectale with DLBCL risk, potentially due to limited sample size. However, the robust association with Ruminococcaceae UCG011 warrants future investigation into its specific mechanisms of action, particularly its potential to modulate these reported butyrate-sensitive pathways in B-cell malignancies.
Alistipes, a recently identified genus of anaerobic bacteria, predominantly colonizes the human gastrointestinal tract but has also been identified in extraintestinal sites including the brain, bloodstream, and gut periphery.53 This bacterial genus demonstrates complex disease-modulating properties, with studies reporting both protective and pathogenic associations. Several studies have reported that Alistipes exerts protective effects against colitis.54 In patients with liver cirrhosis, Alistipes abundance declines progressively from the compensated to the decompensated stage.55, 56 The onset and severity of liver fibrosis have been associated with reduced Alistipes populations. The observed anti-fibrotic effects may be mediated through Alistipes production of propionate and acetate, with the latter exhibiting well-documented anti-inflammatory properties.57, 58 Additionally, emerging evidence indicates complex, context-dependent roles for Alistipes in cancer biology. While elevated Alistipes abundance has demonstrated protective effects against hepatocellular carcinoma (HCC) progression, potentially mediated through the suppression of hepatic T helper 17 cells,59 other studies suggest this genus may promote carcinogenesis in colorectal cancer via IL-6/STAT3 pathway activation.60 Our findings revealed a similarly protective association between Alistipes and DLBCL risk, which may be mediated through immunomodulatory mechanisms analogous to those observed in hepatic malignancies. However, the precise pathways underlying Alistipes–DLBCL interactions require further mechanistic investigation.
Moreover, our MR analysis revealed a significant positive association between Bilophila and the risk of DLBCL. The anaerobic genus Bilophila has been implicated in diverse pathological conditions, including abscesses, appendicitis, colitis, and Parkinson’s disease.61 Studies have demonstrated that Bilophila generates hydrogen sulfide (H2S), a gaseous metabolite that significantly compromises intestinal barrier function. This disruption facilitates direct contact between harmful substances or bacteria and epithelial surfaces, thereby impairing immune responses, activating inflammation, and ultimately promoting colorectal tumorigenesis.62, 63 Furthermore, evidence also suggests that Bilophila exhibits inhibitory effects on butyrate-producing gut microbiota, simultaneously affecting both microbial balance and host physiology.64 Given the established protective role of butyrate against DLBCL, it is plausible that Bilophila may contribute to DLBCL pathogenesis through butyrate depletion mechanisms. However, the precise nature of this relationship remains to be elucidated, as no prior studies have directly investigated Bilophila–DLBCL associations.
The gut microbiota serves as a key biosynthetic organ for circulating metabolites, generating diverse small molecules that systemically regulate host physiology through multiple mechanisms. Emerging evidence links dysregulation of these microbial metabolic pathways to various disease states.65, 66 As previously discussed, elevated levels of butyrate have been associated with reduced lymphoma burden, whereas the loss of butyrate-producing bacteria may diminish these beneficial effects.24 Moreover, this relationship is further supported by observations in NK/T-cell lymphoma patients, who exhibit significantly reduced levels of both butyrate and its primary producer Faecalibacterium prausnitzii. Butyrate is hypothesized to inhibit tumor progression by activating SOCS1 and suppressing the JAK–STAT signaling pathway.67 Building upon our initial findings, we conducted a comprehensive investigation of the causal relationships between gut microbiota-derived metabolites and DLBCL risk. Our analysis identified indolepropionate, a microbial metabolite generated from dietary tryptophan catabolism,68 as a potentially protective factor for DLBCL. This compound demonstrates multifaceted biological activity, including the stimulation of interleukin (IL)-10 secretion from bone marrow-derived macrophages to exert potent anti-inflammatory effects.69 Another study demonstrated that indolepropionate attenuates intestinal inflammation by suppressing interferon gamma (IFN-γ), TNF-α, and IL-1β through its action as an aryl hydrocarbon receptor (AHR) ligand that promotes IL-22 production.68 Indolepropionate has also been reported to exhibit anti-tumor effects by inhibiting epithelial-mesenchymal transition, enhancing anti-tumor immunity through upregulation of both AHR and pregnane X receptor (PXR) pathways, and functioning as a free radical scavenger against oxidative DNA damage induced by carcinogens like free iron and Cr(III).70, 71, 72 The convergence of these anticancer properties with our MR findings suggests its potential relevance to DLBCL prevention and treatment, although the specific mechanisms in lymphoid malignancies remain to be fully elucidated.
In contrast to well-characterized microbial metabolites, the pathological significance of 7-HOCA remains poorly understood. A recent study reported elevated levels of 7-HOCA in patients with hepatitis and liver cancer and demonstrated that 7-HOCA induces DNA damage and promotes tumorigenesis in non-alcoholic fatty liver disease (NAFLD).73 In our MR analyses, 7-HOCA was identified as a potential risk factor for DLBCL, providing a compelling rationale for further investigations into the molecular mechanisms through which 7-HOCA may contribute to lymphomagenesis.
This study has several methodological strengths compared with previous research. We employed a MR design to infer causal relationships between gut microbiota, microbiota-derived metabolites, and DLBCL. Bidirectional MR analyses were conducted using large-scale GWAS datasets to comprehensively evaluate these associations. Single nucleotide polymorphisms were selected as instrumental variables to emulate the random allocation of genetic variants. According to Mendel’s laws, these variants are randomly assigned at conception and are generally independent of environmental confounders. As highlighted by Ference et al.,74 this intrinsic property enables MR to estimate causal effects of exposures on disease outcomes with reduced confounding. By serving as proxies for long-term exposure, SNPs facilitate more robust causal inference. To assess potential horizontal pleiotropy, we applied both MR-Egger regression and MR-PRESSO. Finally, the robustness of our findings was further supported through complementary analyses using BWMR.
Limitations of the study
Despite its strengths, this study has several limitations that warrant consideration. First, the GWAS data on gut microbiota and metabolites were predominantly derived from European cohorts, and the DLBCL dataset also consisted exclusively of individuals of European ancestry. Consequently, our findings may be susceptible to population stratification bias and may not be generalizable to non-European populations. Second, the absence of clinical subtyping data precluded important stratified analyses, including the evaluation of differences according to cell-of-origin classification (germinal center B-cell (GCB) vs non-GCB subtypes) or the presence of gastrointestinal involvement. Finally, the resolution of the gut microbiota data was limited to the genus level, and the metabolite dataset was incomplete, thereby constraining more in-depth mechanistic exploration. Future studies incorporating more comprehensive and higher-resolution datasets are warranted to validate and refine the inferred causal relationships between gut microbiota and DLBCL.
Conclusions
In summary, our findings provide evidence supporting potential causal relationships between gut microbiota, microbiota-derived metabolites, and DLBCL, offering new insights into DLBCL pathogenesis and informing future diagnostic and therapeutic strategies. However, given the limitations of this study, further experimental and clinical investigations are warranted to more comprehensively elucidate the roles of gut microbiota and their metabolites in DLBCL development.
Supplementary data
The supplementary materials are available at https://doi.org/10.5281/zenodo.17047566. The package contains the following files:
Supplementary Fig. 1. The forest plots of the causal effects of gut microbiota and derived metabolites on the risk of DLBCL.
Supplementary Fig. 2. The funnel plots of the causal effects of gut microbiota and derived metabolites on the risk of DLBCL.
Supplementary Fig. 3. The leave-one-out analyses of the causal effects of gut microbiota and derived metabolites on the risk of DLBCL.
Supplementary Fig. 4. The scatter plots of the causal effects of DLBCL on the risk of gut microbiota.
Supplementary Fig. 5. The forest plots of DLBCL on the risk of gut microbiota.
Supplementary Fig. 6. The funnel plots of DLBCL on the risk of gut microbiota.
Supplementary Fig. 7. The leave-one-out analyses of DLBCL on the risk of gut microbiota.
Data Availability Statement
All datasets used in this study are publicly available. Gut microbiota GWAS data were obtained from the MiBioGen consortium (https://mibiogen.gcc.rug.nl), metabolite GWAS data from the TwinsUK and KORA studies annotated via the Human Metabolome Database (https://hmdb.ca), and DLBCL summary statistics from the FinnGen consortium (https://www.finngen.fi/en, release R10, December 2023). The code for Mendelian randomization analyses is available in Zenodo at https://doi.org/10.5281/zenodo.17047705.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.
.jpg)
.jpg)