















Abstract
Background. Reliable toxicological analysis is crucial for accurate forensic and clinical interpretation; however, pre-analytical factors such as handling and storage can significantly alter drug concentrations in postmortem (PM) samples, potentially leading to misinterpretation. Postmortem degradation, influenced by enzymatic and microbial activity, can change drug levels, making it essential to understand drug stability in biological matrices.
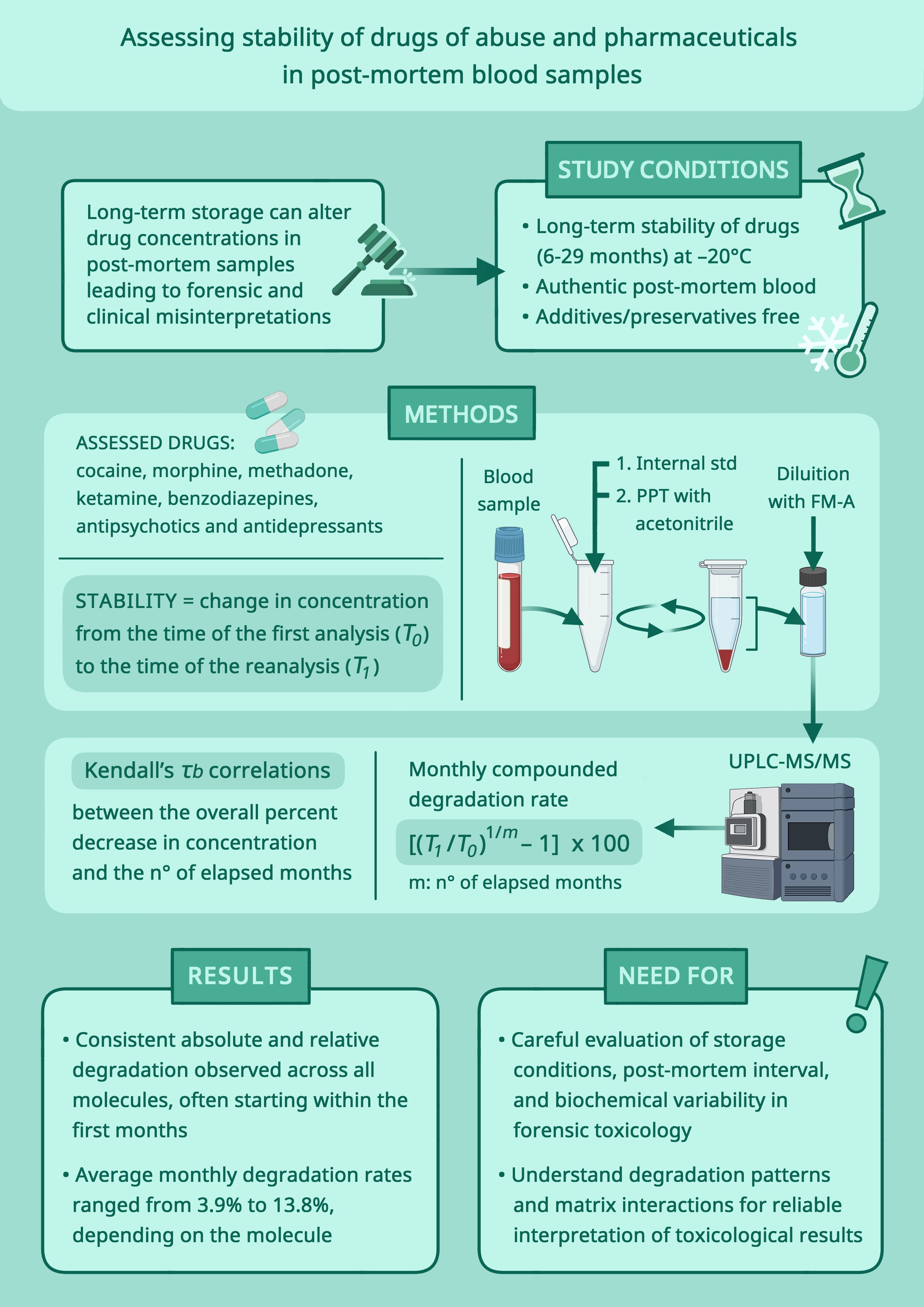
Objectives. This preliminary study investigates the long-term stability of drugs of abuse and psychoactive substances in PM blood samples from drug-related deaths stored at –20°C for 29 months.
Materials and methods. Postmortem blood samples were analyzed using ultra-performance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS) following the routine methodology currently in use in the forensic toxicology laboratory. Stability was assessed by measuring concentration changes between analyses performed shortly after sample collection and reanalyses conducted after 6–29 months. Regression analyses were used to relate percentage variation in concentration to elapsed time.
Results. A strong correlation was found between the percentage reduction in drug concentration and storage time for all tested molecules, including morphine, cocaine, methadone, ketamine, benzodiazepines, antidepressants, antipsychotics, and lidocaine. Regression curve analysis revealed a reduction in concentration beginning within the initial months, with high variability.
Conclusions. The study highlights the significant impact of long-term storage on drug concentrations in PM blood, emphasizing the need for careful consideration of storage intervals when reanalysis of samples is requested for forensic purposes. The findings underscore the importance of understanding degradation patterns for the accurate interpretation of toxicological results in medicolegal investigations.
Key words: forensic toxicology, long-term storage, UPLC-MS/MS, postmortem stability, drug degradation
Background
One of the fundamentals of forensic toxicology is the accurate and reproducible analysis of biological samples. Reliable qualitative and quantitative toxicological analysis is essential for sound toxicological judgment in both clinical and forensic toxicology, as unreliable results may lead to misinterpretations, unwarranted conclusions, or incorrect treatment.1 For this purpose, forensic toxicologists implement rigorous quality control procedures, method validation, and optimization procedures to ensure analytical quality and achieve the highest degree of accuracy.
However, even the most accurate and precise methods reflect drug detection and quantification only at the time of analysis.2, 3 This issue is particularly important when sample re-testing is required for forensic purposes in judicial proceedings. In fact, the majority of “laboratory errors” have been found to originate from the pre-analytical phase rather than from problems related to the analytical process; handling and storage conditions can significantly alter concentrations, especially in samples collected postmortem (PM).4
The degradation of compounds that occurs in PM samples can be caused by several factors, including residual enzymatic or microbial activity or spontaneous hydrolysis.5 Enzymatic activity may degrade drugs into byproducts that are not detected in toxicological analyses or may increase parent drug levels through metabolite hydrolysis. Some microorganisms produce unique nonhuman metabolites, while others are unable to metabolize specific drugs.6 Consequently, assessed PM drug concentrations usually do not reflect drug levels at the time of death, even when analyzing matrices less influenced by PM redistribution, such as peripheral blood.7 Therefore, studying drug stability in biological matrices during storage is of the utmost importance when assessing suspected drug-related crimes.8
Levine and Smith9 were the first to review the stability of compounds of toxicological interest in blood, plasma, serum, urine, oral fluid, and tissue samples. Despite the rising volume of experimental and laboratory research published in forensic toxicology,10, 11 in most studies stability has been investigated using fresh or PM spiked blood samples12, 13, 14, 15 or simulated matrices.16 Fewer experiments have examined the stability of compounds of forensic interest in real samples collected PM.17 The presence of multiple analytes, which is very common in drug-related death cases, can influence the stability of some compounds; however, this issue has rarely been taken into consideration.18
The Guidelines for Sample Collection Postmortem19 recommend the collection of blood in tubes containing both preservatives and no preservatives. The addition of preservatives, such as fluoride at concentrations of up to 2%, to collected matrices has been shown to increase the stability of analytes over time. One of the main reasons for this practice is to prevent PM degradation of molecules, which may occur even under storage conditions. Since the Guidelines19 state that sampling can also be performed without preservatives, it is necessary to understand the stability of these compounds for medico-legal purposes. Nevertheless, in routine forensic casework, most laboratories analyze samples within a short time frame, making the addition of preservatives unnecessary. In this context, the re-testing of samples stored for a prolonged period without preservatives presents greater complexity in terms of interpretation.
Objectives
Our experimental study aimed to investigate the long-term stability of drugs of abuse and psychoactive substances in real, unpreserved PM samples from drug-related deaths stored at –20°C over a period of 29 months.
Materials and methods
Chemicals and reagents
Cocaine, benzoylecgonine, cocaethylene, morphine, codeine, methadone, 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP), opioid analgesics, ketamine, norketamine, and the standards of all included drugs (antipsychotics, antidepressants, benzodiazepines),20 nordiazepam-d5, and ammonium formate were purchased from Sigma-Aldrich® (Steinheim, Germany). Ultra-pure water was obtained by filtration using a Purelab® Chorus 1 Elga system (High Wycombe, UK). Formic acid, acetonitrile, 2-isopropanol, and methanol were purchased from Merck® (Darmstadt, Germany).
Instrumentation
Chromatography was performed using an Acquity UPLC® System (Waters Corporation, Milford, USA) equipped with an Acquity UPLC® HSS C18 column (2.1 × 150 mm, 1.8 µm; Waters Corporation) set at 50°C. The mobile phase consisted of an aqueous solution of 5 mM ammonium formate with 0.1% (v/v) formic acid (A) and acetonitrile with 0.1% (v/v) formic acid (B). The UPLC system was coupled to a Waters triple quadrupole detector (Xevo TQD®) with electrospray ionization (ESI) in positive mode, and data acquisition was carried out in Multiple Reaction Monitoring (MRM) mode. Data acquisition and analysis were performed using MassLynx v. 4.2® software (Waters Corporation), whereas quantitation was performed using the TargetLynx application (Waters Corporation).
Sample collection
The analysis in this study was conducted on 44 PM blood samples collected in 10-mL Vacutainer tubes without any additive or preservative and analyzed for medico-legal purposes at the Laboratory of Forensic Toxicology of the University of Bologna (Italy) between January 2022 and November 2023, according to standardized procedures.21 The cohort comprised 33 men and 11 women. All specimens were stored at −20°C before and after analysis. In May 2024, 1 tube from each sample was reanalyzed under the same instrumental conditions. The time intervals for reanalysis were heterogeneous, ranging from 6 to 29 months.
Sample preparation and analysis
The sample pre-treatment consisted of protein precipitation with ice-cold acetonitrile added to 100 µL of blood. After centrifugation and dilution with mobile phase A, 5 μL was injected into the ultra-performance liquid chromatography−tandem mass spectrometry (UPLC-MS/MS) instrument. The analytical procedure for both the original analyses and the subsequent reanalyses was the same and consisted of a method for the simultaneous detection of drugs of abuse (cannabinoids, cocaine and metabolites, opiates and methadone, amphetamine-like drugs, ketamine) and 68 commonly prescribed antidepressants, benzodiazepines, neuroleptics, and their metabolites in whole blood.20
Statistical analyses
Stability was defined as the ability of a drug concentration to remain unchanged during storage over time. Stability was measured as the change in concentration from the time of the first analysis to the time of reanalysis and was expressed either as the absolute difference in concentration or as the percentage change. The following drugs (or classes of drugs) were assessed: morphine and codeine, cocaine, benzoylecgonine, cocaethylene, methadone, EDDP, ketamine, norketamine, benzodiazepines, antidepressants, antipsychotics, and lidocaine.
For each molecule, we computed the average monthly decrease in concentration by dividing the absolute difference between T0 and T1 by the number of elapsed months. Confidence intervals (CIs) were estimated using nonparametric bootstrapping (1,000 replications) with the bias-corrected and accelerated (BCa) method to account for the small sample size, potential outliers, and heteroskedasticity in the estimates.22 A 95% CI entirely below zero was interpreted as evidence of a statistically significant monthly decrease at the 5% level.
The same analysis was replicated in percentage terms using the monthly compounded degradation rate. This was calculated from the ratio T1/T0 under the assumption that the molecule concentration decreases each month by a constant proportion, consistent with an exponential decay model. This yielded the formula
[(T1/T0)^(1/m) − 1] × 100,
where “m” denotes the number of elapsed months.
While exploratory regressions (e.g., applying square-root or log transformations of time) were initially considered, we refrained from formal modeling due to the small sample sizes (ranging from 4 for norketamine to 19 for benzodiazepines), the lack of repeated measurements or grouping structures, and the absence of theoretical support for a specific degradation law. We therefore prioritized robustness, interpretability, and minimal reliance on distributional assumptions. As a secondary analysis, we computed Kendall’s τb correlations between the overall percentage decrease in concentration and the number of elapsed months. We used Kendall’s τb rather than Spearman’s ρ because it is more robust in small samples and provides a stricter correction for tied ranks in either variable. Given the frequency of ties in elapsed time and the limited sample size, τb offered a more appropriate nonparametric correlation measure for our analysis.23
As this was an exploratory study, no corrections for multiple comparisons were applied. Each molecule was analyzed separately, and no formal comparisons were made across substances. Nevertheless, we acknowledge that multiple hypothesis testing may inflate the type I error rate, and the results should be interpreted accordingly. All analyses were conducted using Stata/SE v. 18 (StataCorp LLC, College Station, USA).
Results
Absolute and relative estimates of average monthly degradation for all tested molecules are reported in Table 1. The results of the T1/T0 analysis for all samples are reported in Supplementary Table 1 and 2.
Discussion
The interpretation of toxicological results in PM samples remains a widely debated topic in forensic toxicology, primarily due to artificial increases or decreases in PM blood concentrations. The main factor responsible for increased concentrations is PM redistribution, a well-known phenomenon describing site- and time-dependent changes in drug levels after death. Drugs bound to tissues can be released from areas of higher concentration, such as the lungs, liver, or stomach contents, and subsequently move along concentration gradients, leading to an artificial rise in PM blood concentrations.
Conversely, drug concentrations may decrease due to tissue uptake, metabolism, or instability and degradation resulting from decomposition or bacterial activity.24 These processes can significantly affect the interpretation of toxicological results and may lead to misjudgment of the toxicological significance of drug concentrations in medicolegal investigations. Besides PM redistribution, stability may also lead to alterations in PM concentrations. In fact, the time between the initial analysis and any subsequent reanalysis can be prolonged, sometimes lasting months or even years, depending on the legal context. Therefore, PM stability is a critical factor that must be systematically evaluated in forensic toxicology. We addressed this issue by investigating concentration changes in drugs of abuse (cocaine and its metabolites, opiates, ketamine and its metabolites, methadone) and pharmaceuticals (benzodiazepines, antidepressants, antipsychotics) in PM blood stored at −20°C without preservatives for up to 29 months.
The analytical results demonstrate absolute and relative reductions in concentration over time for all tested molecules, beginning within the initial months. The results of the secondary analysis (Supplementary Table 1) highlight the limitations of raw time-to-decrease correlations in this context and support the use of normalized degradation rates that explicitly account for time as a key component of the phenomenon under investigation. These rates are not adjusted post hoc but intrinsically incorporate time through their construction formula, which expresses degradation per unit of time under the assumption of constant proportional loss.
This approach is especially appropriate when the time intervals between the 1st and 2nd analyses are highly variable, as in our study (ranging from 6 to 29 months), and when the sample size for each molecule is small, thereby limiting the robustness of traditional regression models. By calculating monthly compounded degradation rates and estimating their uncertainty via bootstrapping, we were able to identify consistent and significant degradation patterns across all molecules, even in the absence of a strong linear or monotonic correlation between elapsed time and percentage decrease.
Morphine and codeine showed an average monthly percentage change of 4.4%, with significant variability among samples. In fact, 5 out of 12 samples demonstrated very good stability, with reductions below 20% at 6, 11, and 19 months, while 3 samples showed reductions greater than 30% starting at 5 months. In the forensic literature, the stability of morphine in real samples has been primarily studied using spiked blank matrices obtained from living subjects.1, 9 In a study performed by Høiseth et al.,25 the stability of opiates in 37 PM blood samples obtained from deaths following heroin intake was investigated. They observed good stability in PM samples (−12% for morphine and −11% for codeine over 4–9 years), which was lower than that observed in samples from living individuals. The results suggest that, for extended storage periods, the use of preservatives for opioids is strongly recommended.
Given the high number of cocaine-positive samples and its metabolites, cocaine, benzoylecgonine, and cocaethylene were studied separately. Similar average monthly percentage changes were observed for all 3 molecules (4.4%, 5.5%, and 3.9%, respectively). Studies assessing the stability of cocaine in real casework include only a small number of cases. Kiszka et al.26 tested the stability of cocaine for up to 90 days in PM blood obtained from 5 corpses. They observed degradation of up to 7% of the initial concentration in frozen samples without preservatives, whereas samples containing preservatives (NaF, CH3COOH, and NaF + CH3COOH) showed good stability.26 Although stability in real samples with preservatives was not studied over longer time intervals, these findings suggest that preservatives slow degradation and should be considered during sampling, especially when reanalysis is performed after months or years. Methadone showed an average monthly percentage change of 5.9%. Notably, several samples demonstrated significant decreases even after short time intervals (5–10 months). Methadone has shown very good stability in clinical plasma samples27; however, despite being increasingly detected in drug-related deaths, particularly in cases of co-administration, there is a lack of stability studies on methadone in PM samples. Benzodiazepines (6.5%), antidepressants (7.4%), and antipsychotics (5.4%) exhibited significant reductions over time, even during early periods. Melo et al.28 conducted an experimental study on unpreserved PM blood spiked with benzodiazepines, observing reductions of up to 13% at 6 months. The wide variability and marked degradation observed in our study may be attributable to the longer study intervals and the heterogeneity of the drugs examined. Additionally, very high variability was observed within this class of molecules, with concentration reductions ranging from 26% to 92% at 28 months. Similar results were observed for antidepressants (−60.4%), whereas antipsychotics showed greater stability, albeit with high inter-sample variability. For ketamine (13.8%) and lidocaine (5.4%), the limited sample size precluded firm conclusions, although significant long-term reductions in stability were observed for these molecules.
In general, although a strong relationship between percentage reduction and elapsed time was observed for all molecules, it is not possible to formulate a model for back-calculating the time elapsed since the initial analysis, as significant inter-sample variability was observed that cannot be predicted using a single model. Therefore, the results should be interpreted in terms of likely concentration reductions upon reanalysis, with careful consideration of quantitative findings.
The differences in stability among samples may be attributed to several factors. First, the initial concentrations of the molecules in the starting samples varied, which may influence the degradation rate.1 Additionally, heterogeneity in the composition of PM samples may account for the fact that PM blood typically exhibits a lower pH compared with blood collected from living individuals, thereby promoting hydrolysis. This pH may vary depending on the PM interval (PMI), and its effect on analytes differs according to the molecular structure of the drug.4
Moreover, biological sex may represent an additional source of variability. It is well established in forensic toxicology that sex influences drug pharmacokinetics and pharmacodynamics; however, little is known about the influence of sex on the decrease in drug concentrations in PM samples.29 Therefore, future studies should also consider sex as a variable, both when defining PM redistribution as a function of PMI and when assessing stability.
Regrettably, given the limited number of cases included in this preliminary analysis, subgroup analyses were precluded due to insufficient statistical power. Finally, based on the current literature, sample stability in tubes containing preservatives appears to be superior and more reliable over extended periods. However, in the absence of comparative experimental studies on real-world samples employing a study design analogous to the present investigation, this hypothesis requires further experimental validation.
Limitations
This study has several unavoidable limitations in the forensic context. Primarily, the subjects from whom the samples were collected had varying PMIs, potentially leading to differential degradation patterns. Consequently, future research could benefit from conducting multiple analyses on single-sample aliquots. However, this approach would introduce the confounding effect of freeze–thaw stability. Furthermore, in this study, most samples tested positive for multiple drugs, as is commonly observed in real forensic casework, and there may be unknown interactions that warrant further investigation. In addition, the influence of the subject’s sex on the decrease in drug concentrations in PM samples warrants further examination. This analysis was not feasible in the current preliminary study due to the limited number of cases included.
Conclusions
The lack of stability observed in this preliminary study highlights the critical importance of considering storage conditions and biochemical variations when evaluating re-testing results in forensic investigations. Potential interactions between different compounds within the blood matrix may affect the stability of individual target analytes, thereby complicating the interpretation of toxicological results. This underscores the necessity for comprehensive analytical strategies that account for PM alterations to ensure reliable and legally defensible results.
Supplementary files
The supplementary materials are available at https://doi.org/10.5281/zenodo.15737109. The package contains the following files:
Supplementary Table 1. The results of the analysis at T1/T0 for all samples are reported.
Supplementary Table 2. Kendall’s τb correlations between the overall percent decrease in molecule concentration and the number of elapsed months.
Data Availability Statement
The datasets supporting the findings of the current study are openly available in Zenodo at https://doi.org/10.5281/zenodo.15082513.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.