Abstract
Background. High levels of PM2.5 air pollution pose serious health risks, especially in rapidly urbanizing areas. While its effects on organs such as the heart and lungs are well documented, its effects on the cornea remain less well understood. Emerging evidence links PM2.5 exposure to corneal damage through processes such as autophagy, inflammation, and oxidative stress; however, the precise molecular pathways remain largely unknown.
Objectives. This study aimed to identify key genes and signaling pathways in PM2.5-exposed human corneal epithelial cells (HCECs) using RNA sequencing and bioinformatics analysis.
Materials and methods. Human corneal epithelial cells were cultured and exposed to 25 μg/mL PM2.5 for 24 h. High-throughput sequencing was performed after total RNA extraction and library construction for mRNA and microRNA (miRNA). Clean reads were mapped to the reference genome after filtering out low-quality reads. The Differential Expression Sequencing 2 (DESeq2) R package was used to identify differentially expressed (DE) mRNAs and miRNAs with a fold change ≥2 or ≤0.5 and a false discovery rate (FDR) ≤ 0.001.. Bioinformatics analyses included hierarchical clustering, protein–protein interaction network construction, target gene prediction, and Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment.
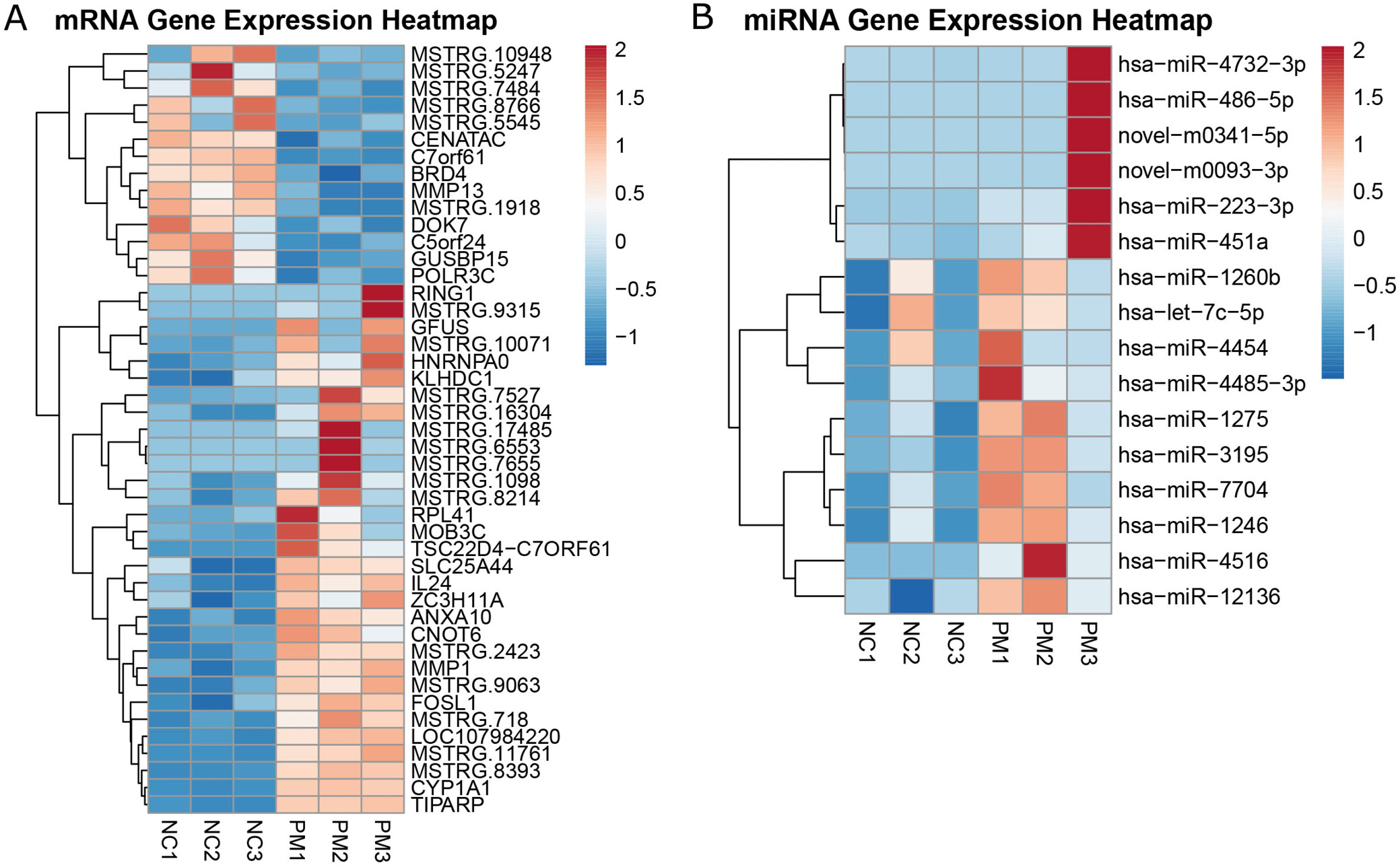
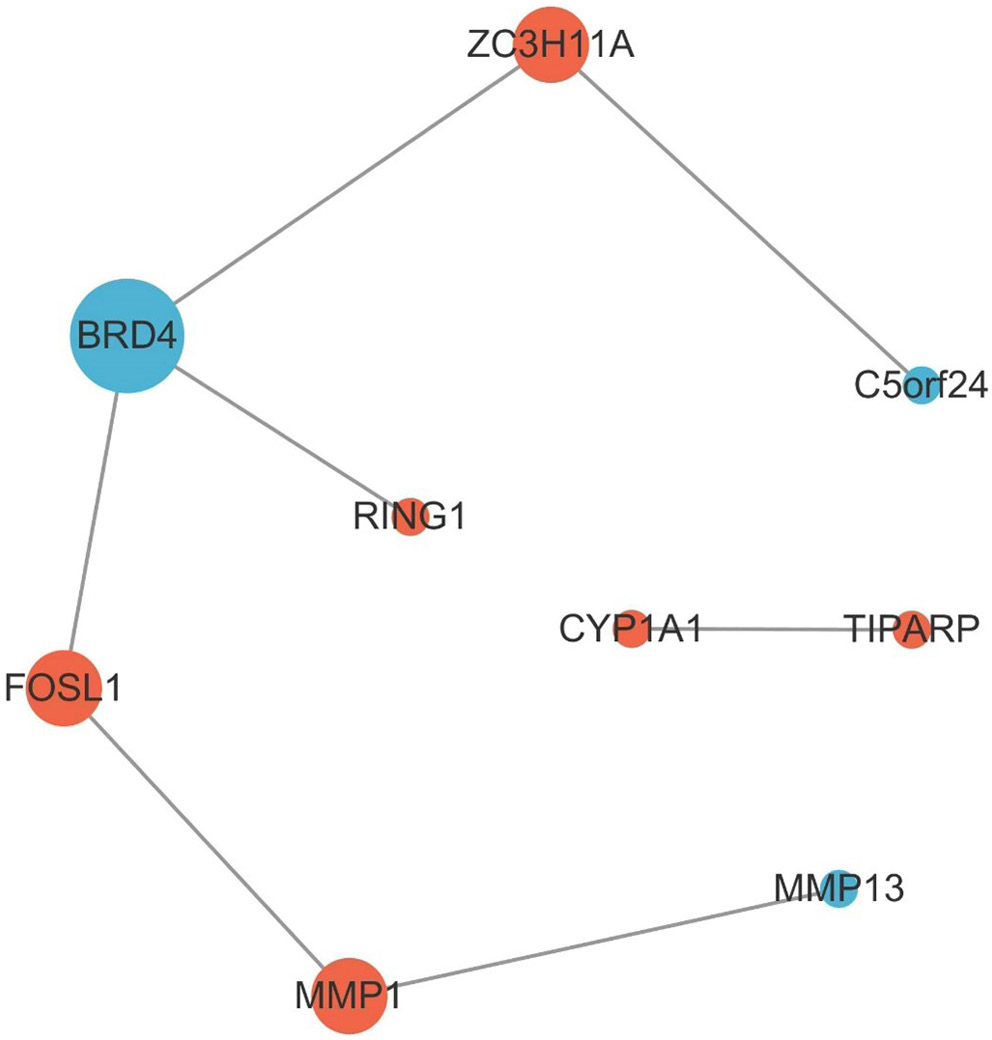
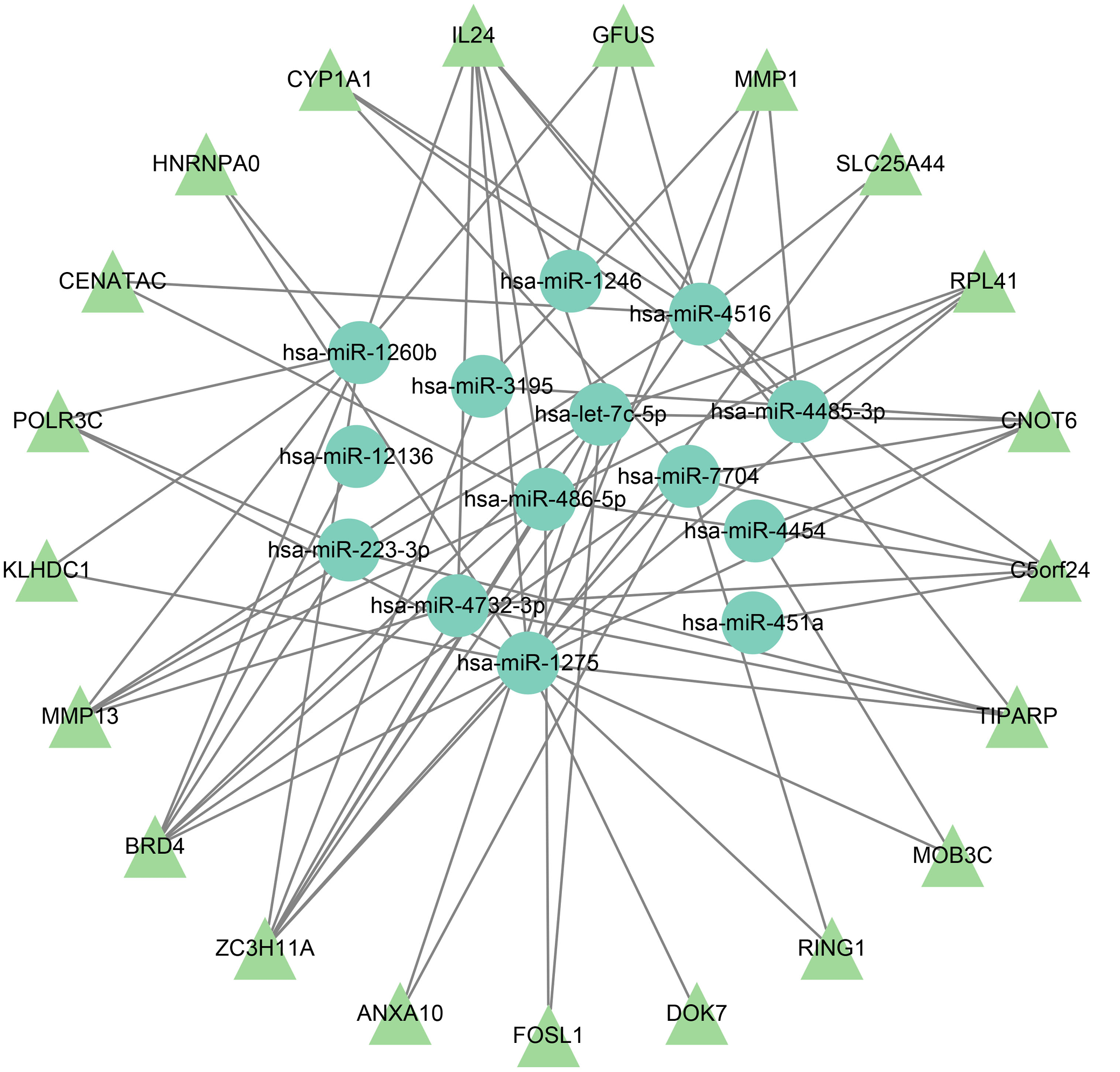
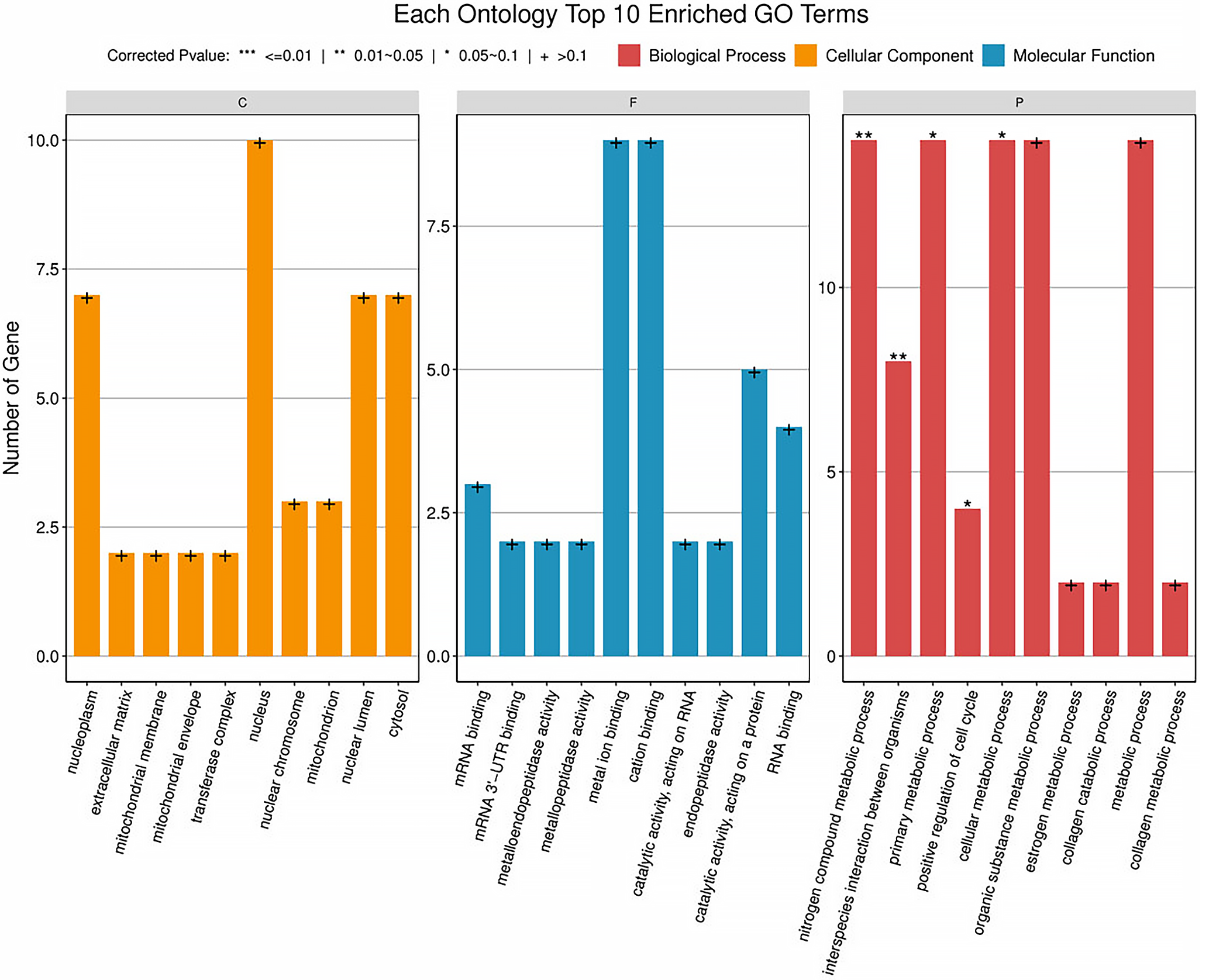
Results. The analysis identified 45 DE mRNAs, including 14 upregulated and 31 downregulated transcripts, along with 16 upregulated miRNAs. A gene interaction network was constructed comprising nine mRNAs (6 upregulated and 3 downregulated), while a combined miRNA–mRNA network included 14 miRNAs and 21 mRNAs, forming 73 interaction pairs. Functional enrichment analysis of these genes revealed 30 significantly enriched GO terms, as well as 27 KEGG signaling pathways.
Conclusions. This study constructed regulatory networks and identified genes DE in the corneal response to PM2.5 exposure, particularly those involved in autophagy, inflammatory responses, and oxidative stress–related pathways. These results lay the groundwork for further research into the effects of PM2.5 on ocular surface health and provide insights into the molecular mechanisms underlying PM2.5-induced damage to human corneal epithelial cells, potentially guiding the development of targeted diagnostics or therapies to mitigate ocular surface injury caused by PM2.5.
Key words: PM2.5, bioinformatics analysis, human corneal epithelial cells
Background
Air pollution is a well-established global public health concern.1 Among its various components, fine particulate matter (PM2.5) is considered particularly hazardous.2, 3 In developing nations undergoing rapid industrialization and urban expansion, such as China, PM2.5 levels have reached critical thresholds; for instance, the annual average PM2.5 concentration in eastern China exceeded 80 μg/m3 in 2010.4, 5 Exposure to PM2.5 has been widely linked to elevated risks of cancer, respiratory and cardiovascular diseases, and increased mortality.6, 7, 8, 9, 10
Despite these findings, its effects on the ocular surface, particularly the cornea, which is directly exposed to airborne pollutants, have received less attention. Xiang et al. emphasized the harmful effects of PM2.5 on ocular health.11 Recent studies have suggested that PM2.5 may contribute to corneal epithelial damage through mechanisms such as oxidative stress, autophagy dysregulation, and pro-inflammatory responses.12, 13, 14, 15, 16 However, the precise molecular mechanisms involved in these corneal responses remain largely unknown, and whether they mirror those observed in other organs is still unclear.
Objectives
Through comprehensive bioinformatics analysis, this study aims to clarify the key genes and signaling pathways implicated in the effects of PM2.5 on the cornea.
Materials and methods
Culture and exposure of human corneal epithelial cells to PM2.5
Human corneal epithelial cells (HCECs) were provided by the Shandong Eye Institute, Shandong Provincial Key Laboratory of Ophthalmology, Qingdao, China. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F12 medium supplemented with 1% penicillin–streptomycin and 10% fetal bovine serum (FBS). The medium was changed every 2 days, and the cultures were maintained at 37°C in an atmosphere containing 5% CO2.17, 18, 19 Cells were seeded in 6-well plates and cultured for 24–48 h in regular growth medium. The cells were then rinsed with phosphate-buffered saline (PBS) and incubated for 24 h in either control medium or a PM2.5 solution at a concentration of 25 μg/mL. The PM2.5 used was SRM 1648a (National Institute of Standards and Technology (NIST), Gaithersburg, USA), which was suspended in sterile PBS and sonicated for 30 min in a water-bath sonicator (US-220; Zhongke Scientific Instrument, Beijing, China) to ensure even dispersion before application. Each treatment was replicated 3 times for subsequent RNA extraction. Three biological replicates were used for both mRNA and miRNA sequencing analyses. The cells used for all experiments were between passages 2 and 5. The PM2.5 employed was the urban particulate matter reference standard (SRM 1648a), sourced from the NIST and representative of urban environments.
This study did not involve any human participants or animal experiments. Only commercially available human corneal epithelial cell lines were used. Therefore, ethical approval was not required.
Library preparation
RNA library preparation
Total RNA from 2 distinct groups was qualified and quantified using a bioanalyzer (Agilent 2100 Bioanalyzer; Agilent Technologies, Santa Clara, USA) before being randomly fragmented. These fragments then served as templates for 1st-strand cDNA synthesis, followed by 2nd-strand synthesis. The fragments were then purified using the QIAquick polymerase chain reaction (PCR) kit (Qiagen, Hilden, Germany), eluted with elution buffer, end-repaired, adenylated by the addition of a single ‘A’ base, and ligated to sequencing adapters. The uracil-N-glycosylase (UNG) enzyme (Thermo Fisher Scientific, Waltham, USA) was then used to degrade the 2nd strand. Following fragment size selection by agarose gel electrophoresis, polymerase chain reaction (PCR) amplification was performed to generate the sequencing library.
microRNA library preparation
RNA concentrations in 6 samples were determined using a bioanalyzer (Agilent 2100 Bioanalyzer; Agilent Technologies). RNAs of different fragment sizes were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and bands ranging from 18 to 30 nucleotides were excised to recover small RNAs. According to the manufacturer’s instructions for T4 RNA ligase (Thermo Fisher Scientific), these RNAs were ligated to 5′ and 3′ adapters before being reverse-transcribed into cDNA molecules. To complete library construction, the PCR products were purified by PAGE and dissolved in ethidium bromide (EB) solution.
RNA-seq method
Raw sequencing reads were subjected to quality control using FastQC v. 0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to evaluate metrics such as per-base sequence quality, GC content (the molar ratio of guanine + cytosine bases in DNA), and adapter contamination. Trimmomatic v. 0.3920 (http://www.usadellab.org/cms/?page=trimmomatic) was employed to remove adapter sequences and low-quality bases, using parameters: ILLUMINACLIP:2:30:10, SLIDINGWINDOW:4:20, and MINLEN:36. Cleaned reads were aligned to the human reference genome (GRCh38/hg38) using HISAT2 v. 2.2.1 (https://daehwankimlab.github.io/hisat2/),21 a splice-aware aligner. The resulting files of sequence alignment map (SAM) were converted to sorted binary alignment map (BAM) files using SAMtools v. 1.10 (https://www.htslib.org/doc/1.10/samtools.html).22 Subsequently, transcript assembly and quantification were performed with StringTie v. 2.2.1 (https://ccb.jhu.edu/software/stringtie/).23 For each sample, transcript abundance was calculated based on the reference gene annotation (GTF) file, and novel transcript isoforms were also predicted. Expression levels were quantified in fragments per kilobase of transcript per million mapped reads (FPKM). Gene-level count matrices were generated using the prepDE.py script provided with StringTie. Differential gene expression analysis was conducted using the DESeq224 package v. 1.38.3 in R v. 4.2.2 (R Foundation for Statistical Computing, Vienna, Austria). Raw count data were normalized, and statistical testing was performed using the Wald test with Benjamini–Hochberg correction for multiple comparisons. Genes with an adjusted p < 0.050 and |log2 fold change| ≥1 were considered significantly differentially expressed (DE). To interpret the biological significance of the DE genes, Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed using the clusterProfiler25 package (v. 4.6.2) in R. Enrichment results were visualized with dot plots and pathway diagrams to identify biological processes and signaling pathways significantly associated with DE genes.
Results
Analysis of 6 samples from both the PM2.5 group and the negative control group revealed 45 DE mRNAs (Figure 1A and Supplementary Table 1), including 14 upregulated and 31 downregulated transcripts. Similarly, analysis of the same sample set identified 16 DE miRNAs, all of which were upregulated (Figure 1B, Supplementary Table 2). Using Cytoscape software (URL: https://cytoscape.org/), an interaction network of DE mRNAs was constructed, comprising 9 nodes and 7 edges (Figure 2). This network included 6 upregulated and 3 downregulated genes. These 9 mRNAs were selected for downstream analysis based on their centrality and connectivity within the protein–protein interaction (PPI) network, indicating potential roles as regulatory hubs.
The interaction network included 35 DE genes, consisting of 14 miRNAs and 21 mRNAs, with 73 interaction pairs identified (Figure 3). Key genes in the miRNA–mRNA network were selected based on overlap in target prediction, consistency of differential expression, and biological relevance to pathways identified through enrichment analysis.
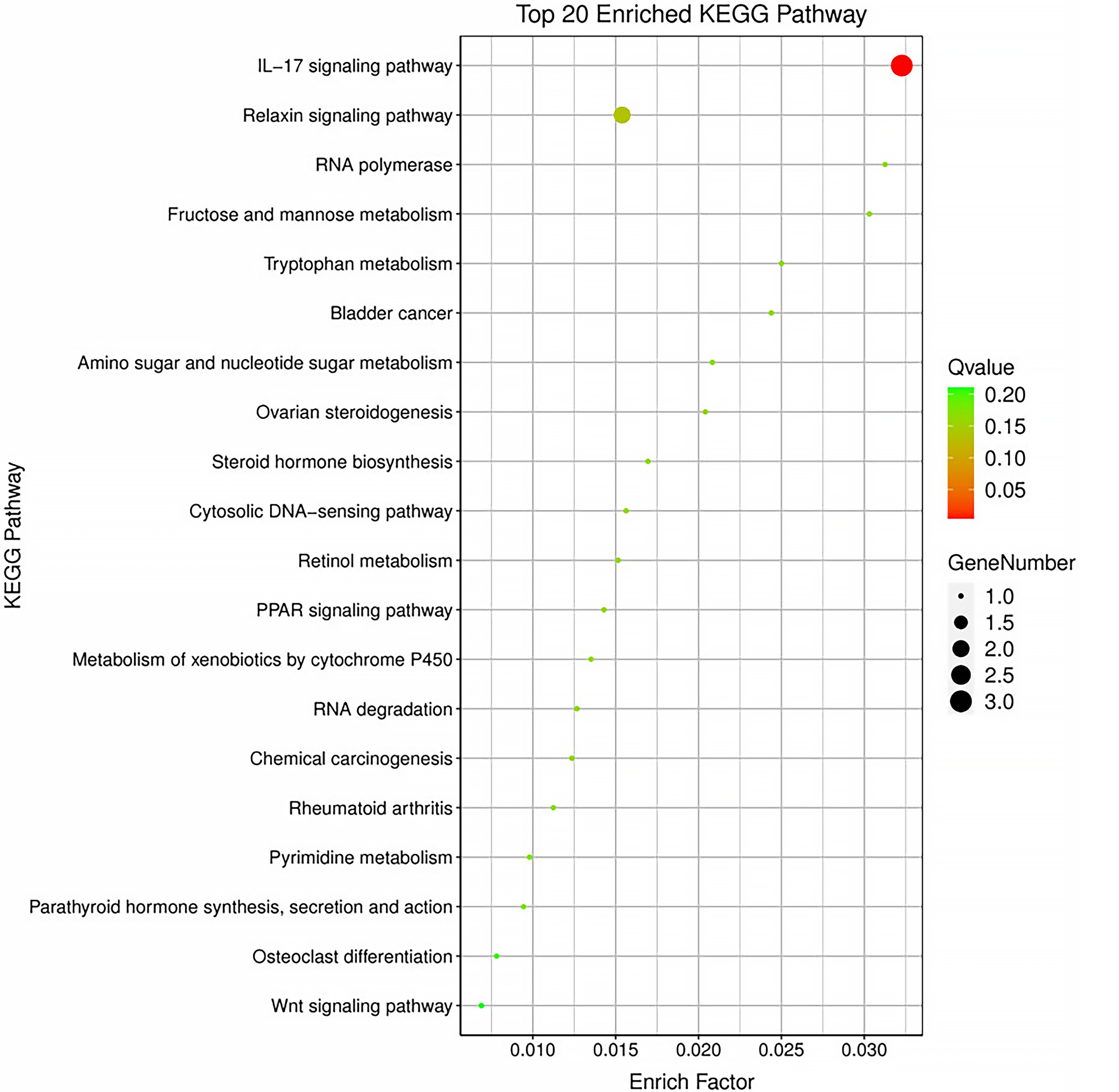
Functional enrichment analysis was performed on the 9 DE mRNAs from the network, revealing enrichment in 30 GO terms, segmented into 3 categories: biological process (BP), cellular component (CC), and molecular function (MF). The top 10 entries from each category are shown in Figure 4 and Supplementary Table 3. Additionally, 27 signaling pathways were identified through KEGG analysis. The top 20 entries from each category are shown in Figure 5 and Supplementary Table 4.
Discussion
PM2.5 is a complex mixture consisting of various sources, such as automobile exhaust, combustion smoke, soil, and cooking fumes. Its constituents include metals, salts, volatile organic compounds, hydrocarbons, and endotoxins.26 Owing to this intricate composition, the impact of PM2.5 on human health is multifaceted. However, its ocular effects, particularly on the cornea, remain understudied. This study aims to explore the influence of PM2.5 on the cornea and to identify key genes and signaling pathways involved.
We identified several novel genes and miRNA candidates not previously associated with PM2.5-induced ocular damage. Our study found that several genes in HCECs exhibited differential expression after exposure to PM2.5. These include BRD4, FOSL1, and CYP1A1, which appear to play central roles in regulating oxidative stress, autophagy, and inflammation in the corneal epithelium. BRD4, a member of the bromodomain and extraterminal protein family, recognizes and binds to acetylated lysine residues on histone tails.27 It is implicated in the regulation of various inflammatory cytokines, including interleukin (IL)-1β, IL-6, IL-8, and IL-9, and functions as a transcriptional regulator that modulates autophagy and oxidative stress via lysosomal pathways.28, 29, 30
FOSL1, a component of the AP-1 transcription factor complex, has been reported to be upregulated following PM2.5 exposure in lung epithelial cells and may contribute to autophagy inhibition, inflammation, and oxidative stress.31, 32 Studies have suggested that FOSL1 can promote apoptosis and inflammation through the AMPK signaling pathway,33 and that its inhibition leads to reduced pro-inflammatory cytokine expression.34, 35 FOSL1 also interacts with the Nrf2 pathway, a key regulator of oxidative stress.36, 37, 38
Cytochrome P450 enzymes (CYPs) are essential for the metabolism of various substances, including heavy metals and drugs.39 CYP1A1, a cytochrome P450 enzyme, has been shown to be upregulated in corneal epithelial cells following exposure to airborne pollutants such as cigarette smoke and house dust.11, 40 It participates in xenobiotic metabolism and is regulated by the aryl hydrocarbon receptor (AhR) signaling pathway.39, 40, 41, 42, 43, 44, 45 Suppression of CYP1A1 has been linked to reduced reactive oxygen species (ROS) production and oxidative stress.45 Its co-regulation with TIPARP via AhR signaling underscores its potential importance in pollutant-induced ocular responses
– In addition, we identified 16 DE miRNAs. Among these, several novel miRNA–mRNA interactions potentially relevant to corneal responses to PM2.5 were identified:
– miR-1275, predicted to target BRD4 and TIPARP, may regulate autophagy and ROS generation and inflammation when inhibited.46, 47, 48
– miR-223-3p, also targeting BRD4 and TIPARP, has been linked to the modulation of autophagy and oxidative stress in both ocular and systemic contexts.49, 50, 51, 52
– miR-486-5p, targeting BRD4 and FOSL1, was shown to induce autophagy upon inhibition and reduce inflammation and oxidative damage.53, 54, 55
– miR-4516, associated with CYP1A1 and TIPARP, may regulate autophagic responses to metal components in PM.56
– miR-4454 expression is elevated in PM-exposed populations, although its ocular mechanism is unclear.57
– let-7c-5p, which may regulate BRD4 and FOSL1, has been implicated in abnormal autophagy in lens epithelial cells.58
Gene Ontology analysis and KEGG signaling pathway enrichment were performed for the DE mRNAs. The enriched BP terms were mainly related to nitrogen compound metabolic processes and primary metabolic processes. The enriched CC terms were primarily associated with the nucleoplasm, extracellular matrix (ECM), and mitochondrial membrane. The enriched MF terms were mainly related to enzyme activation or binding, such as metalloendopeptidase activity, metallopeptidase activity, and metal ion binding. The enriched KEGG pathways included the IL-17 signaling pathway, the peroxisome proliferator-activated receptors (PPAR) signaling pathway, tryptophan metabolism, and the metabolism of xenobiotics by cytochrome P450. Interleukin 17 is considered to play a role in regulating corneal inflammatory responses,59 and the PPAR signaling pathway is thought to be involved in oxidative stress and autophagy.60
These enriched pathways, particularly IL-17 and PPAR signaling, further underscore the mechanistic links between PM2.5 exposure and oxidative stress, inflammation, and autophagy in the cornea.
Limitations
There are several limitations to this study. First, the fine particulate matter used, SRM 1648a, was not directly collected from ambient air but is a reference material. Its composition therefore differs somewhat from that of actual environmental PM. However, its main components, such as various metals, polycyclic aromatic hydrocarbons, and polychlorinated biphenyl homologues, are similar to those typically adsorbed onto atmospheric PM. Second, the DE RNAs identified in this study require further validation. Despite these limitations, our work provides a foundational transcriptomic dataset and identifies several novel genes, pathways, and miRNA–mRNA interactions that may mediate PM2.5-induced corneal damage, thereby laying the groundwork for further elucidation of the underlying damage mechanisms.
Conclusions
Through bioinformatics analysis, we have begun to elucidate genes and signaling pathways responsive to PM2.5-induced damage and have constructed regulatory networks that may underlie these processes. These findings provide new insights into the impact of PM2.5 on ocular health and serve as a platform for future research in this field, potentially guiding the development of targeted diagnostics or therapies to mitigate PM2.5-induced ocular surface damage.
Supplementary files
The supplementary materials are available at https://doi.org/10.5281/zenodo.15245015. The package contains the following files:
Supplementary Table 1. Statistical analysis data of mRNA between the NC group and the PM2.5 group.
Supplementary Table 2. Statistical analysis data of miRNA between the NC group and the PM2.5 group.
Supplementary Table 3. GO analysis of DE mRNAs.
Supplementary Table 4. KEGG signaling pathways of DE mRNAs.
Data Availability Statement
The datasets generated and analyzed during the current study are available in the NCBI repository. The accession number for these SRA data is PRJNA1233430, and its release date is June 30, 2025.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.