















Abstract
Background. Intervertebral disc degeneration (IDD) is the primary cause of lower back pain. Transient receptor potential canonical 3 (TRPC3) is a nonselective cation channel permeable to Ca2+.
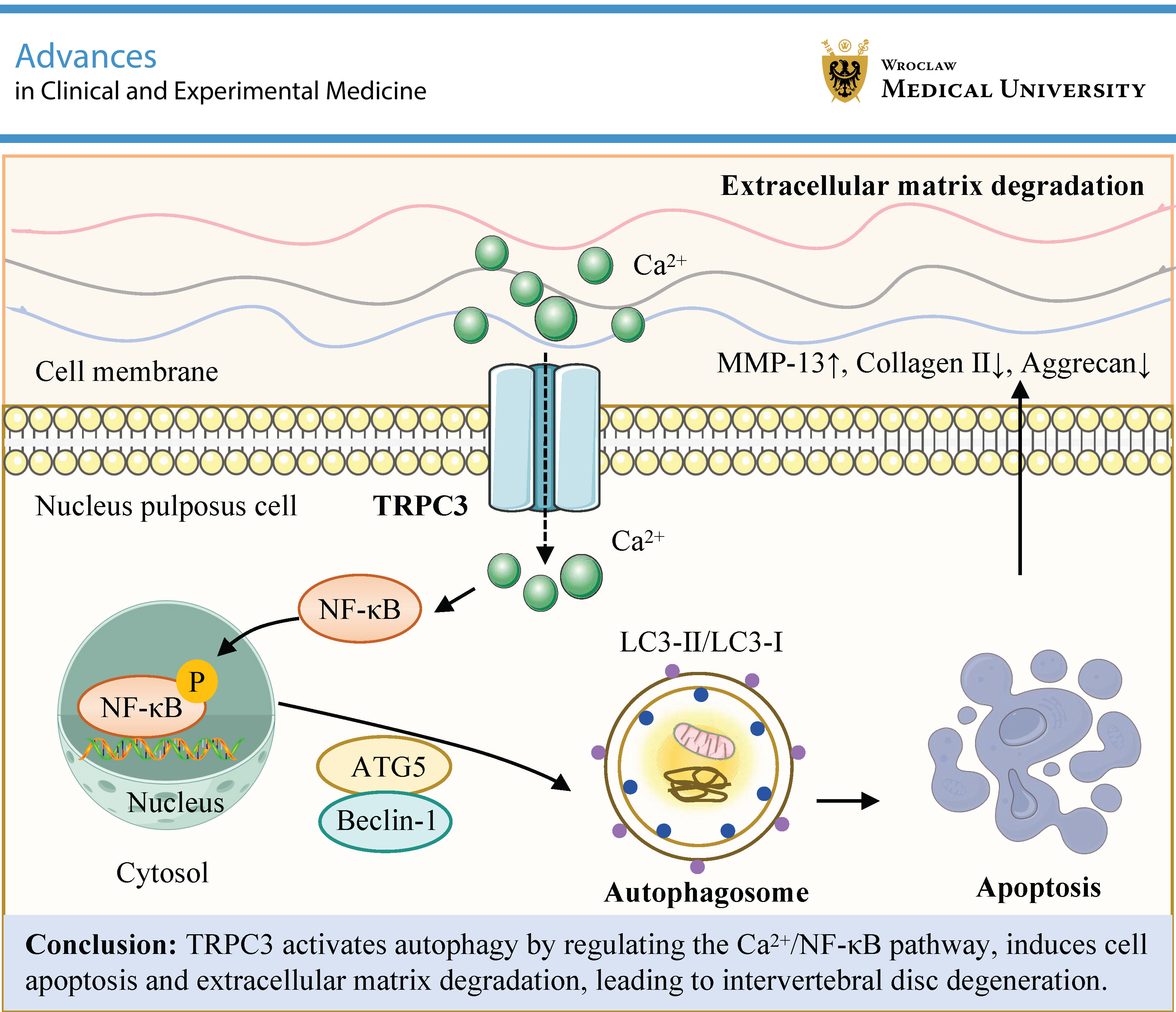
Objectives. This study explores the mechanisms by which the TRPC3-mediated Ca2+/nuclear factor kappa B (NF-κB) pathway regulates autophagy in IDD.
Materials and methods. An IDD rat model was established using the annulus fibrosus puncture method and was treated with local intraspinal injection of adeno-associated virus (AAV)-shRNA targeting TRPC3. Primary human nucleus pulposus cells (NPCs) were transfected with TRPC3 siRNA and subsequently treated with pyrrolidine dithiocarbamate (PDTC; an NF-κB inhibitor), rapamycin (RAPA), or 3-methyladenine (3-MA), respectively. Micro-computed tomography (micro-CT), hematoxylin and eosin (H&E) staining, immunohistochemistry, western blotting, transmission electron microscopy (TEM), and flow cytometry were performed.
Results. TRPC3 expression was significantly increased in IDD rats (p < 0.05). TRPC3 shRNA ameliorated histopathological damage in IDD rats and promoted the expression of autophagy-related protein 5 (ATG5), Beclin-1, and LC3-II (all p < 0.05). In vitro, interleukin-1 beta (IL-1β) increased Ca2+ levels, siRNA TRPC3 reduced them, and PDTC further decreased them (p < 0.05). In addition, siRNA TRPC3 increased the expression of ATG5, Beclin-1, and the LC3-II/LC3-I ratio and inhibited phosphorylation of p-NF-κB p65 in NPCs (p < 0.05). Transmission electron microscopy and flow cytometry showed that siRNA TRPC3-induced autophagy promoted apoptosis in NPCs (p < 0.05). Furthermore, siRNA TRPC3 increased the levels of aggrecan and collagen II and decreased matrix metalloproteinase-13 (MMP-13) expression (p < 0.05).
Conclusions. TRPC3 exacerbates IDD by inhibiting protective autophagy via activation of the Ca2+/NF-κB signaling pathway. Knockdown of TRPC3 promotes autophagy, which in turn influences NPC apoptosis and extracellular matrix (ECM) metabolism. This study offers potential novel strategies for IDD prevention and treatment.
Key words: autophagy, NF-κB, intervertebral disc degeneration, TRPC3, calcium ions influx
Background
Low back pain (LBP) is the most common chronic musculoskeletal condition affecting adults and has emerged as a leading cause of global disability, profoundly impacting patients’ work capacity and daily life.1 Research indicates that 60–80% of individuals experience symptoms of LBP, with approx. 10% of patients becoming disabled.2, 3, 4 Currently, approx. 630 million people worldwide suffer from neck pain and LBP. Intervertebral disc degeneration (IDD) is the primary cause of LBP and disc-related disorders, such as herniation and spinal stenosis.5 Studies have revealed that approx. 40% of low back and leg pain cases are attributable to IDD.6 The prevalence of IDD is steadily increasing across all age groups, posing significant challenges for medical care and society.7 Although IDD is particularly prevalent among the elderly population, a shift toward younger patients has been observed in recent years, likely due to changes in lifestyle and habits.8 Intervertebral disc degeneration is a progressive, multifactorial condition characterized by biomechanical, structural, and biological changes in disc tissue induced by various factors, ultimately leading to the loss of disc integrity and function. These changes include annulus fibrosus rupture, nucleus pulposus (NP) herniation, extracellular matrix (ECM) degradation, reduced disc height, and compression of the spinal cord and nerve roots, ultimately resulting in lower back and leg pain.9 Recent research suggests that IDD is a complex disease resulting from the interaction of multiple factors, including aging, genetic predisposition, mechanical stress, inflammation, oxidative stress, metabolic dysfunction, environmental factors, and autophagy.5, 10, 11, 12 Furthermore, IDD is characterized by degeneration of the intervertebral disc ECM, leading to reduced biomechanical integrity and pain.13 Among these factors, matrix metalloproteinases (MMPs) are key contributors to ECM degradation in IDD. Previous studies have shown that bovine bone grafts can contribute to intervertebral disc repair by supporting cell attachment, proliferation, and matrix synthesis.14 Recent studies have also demonstrated that inhibition of matrix degradation can reduce mitochondrial damage, inhibit cell apoptosis and senescence, and significantly delay the progression of IDD.15 However, the pathogenesis of IDD remains a major challenge in clinical practice. Therefore, it is crucial to further elucidate the mechanisms underlying IDD and identify reliable therapeutic targets to improve ECM metabolic imbalance and ultimately reverse the progression of IDD.
As an intracellular recycling process, autophagy degrades cytoplasmic components via lysosomes and plays a vital role in cellular self-degradation and recycling.16 This process is essential for maintaining metabolic homeostasis. It has been reported that autophagy alleviates osteoarthritis by regulating ECM metabolism.17 Recent studies have increasingly demonstrated that autophagy plays a crucial role in IDD.18, 19 Cheng et al. has found that regulation of chaperone-mediated autophagy can effectively delay the progression of IDD in an inflammatory environment.20 However, the role of autophagy in IDD remains controversial because of its dual effects. Some studies have reported that the expression levels of autophagy-associated genes are significantly higher in IDD than in healthy discs.21 In addition, research has indicated that appropriate activation of autophagy can protect nucleus pulposus cells (NPCs), which are the core cellular components of the intervertebral disc, from pressure-induced damage. In contrast, impairment of autophagic function can lead to apoptosis of NPCs, thereby further accelerating the progression of IDD.22, 23
Transient receptor potential (TRP) channels are known for their involvement in sensory processes, such as temperature and pain perception, as well as in the regulation of cellular calcium homeostasis. Transient receptor potential canonical 3 (TRPC3) is a Ca2+-permeable, nonselective cation channel that plays vital roles in a wide range of cellular physiological processes. TRPC3 facilitates calcium entry, thereby enabling cells to regulate gene expression, as well as cell growth and differentiation. Notably, TRPC3 has been implicated in various physiological and pathological conditions, including cardiovascular diseases, neurological disorders, and cancer. In the context of IDD, the specific mechanisms of TRPC3 remain controversial. Some studies suggest that TRPC3 is involved in cytosolic Ca2+ elevation, activation of nuclear factor kappa B (NF-κB), and cytokine upregulation.24 Notably, research has shown that increased TRPC3 expression can lead to elevated intracellular Ca2+ concentrations and is associated with decreased bone mass.25 Abnormal TRPC3 channel activity may promote bone resorption, reduce bone density, and mediate the Ca2+/NF-κB signaling pathway.26 Calcium (Ca2+) is one of the most abundant and important signaling molecules in the human body. In tissues such as intervertebral discs, bone, and cartilage, elevated intracellular Ca2+ concentrations activate Ca2+ signaling pathways, which in turn regulate gene expression and protein synthesis, thereby influencing changes in the microenvironment of intervertebral disc tissue. Previous investigations have demonstrated the critical role of Ca2+ signaling in intervertebral disc tissues. Intervertebral disc degeneration leads to alterations in the osmotic pressure of intervertebral disc tissue, resulting in activation of the Ca2+ signaling pathway. Intervertebral disc cells regulate gene expression and protein synthesis by increasing intracellular Ca2+ concentrations. Furthermore, Ca2+ acts as a second messenger in the human body, activating downstream signaling pathways. The role of the NF-κB signaling pathway in IDD has been widely recognized; however, its upstream regulatory mechanisms are diverse. Evidence suggests that calcium/calmodulin-dependent protein kinase II, in conjunction with interleukin-1 receptor-associated kinase 1 (IRAK1), plays a critical role in the phosphorylation processes that activate NF-κB.27 When activated, the NF-κB pathway triggers cell apoptosis and ECM degradation, thereby promoting IDD.28 In addition, the NF-κB pathway promotes autophagy in various diseases. Melatonin has been shown to induce autophagy via the NF-κB signaling pathway, thereby preventing ECM degeneration in intervertebral disc cells.29 Furthermore, through the miR-139-3p/CXCR4/NF-κB axis, lncRNA H19 enhances autophagy and apoptosis in NPCs, thereby aggravating IDD.30 However, there are currently no reports on whether TRPC3 can activate autophagy to regulate IDD via the Ca2+/NF-κB pathway. Moreover, whether TRPC3-induced NF-κB activation through Ca2+ influx exerts synergistic effects on apoptosis and ECM degradation remains to be further elucidated.
Objectives
This study aims to investigate the role and underlying mechanisms by which the TRPC3-mediated Ca2+/NF-κB pathway inhibits autophagy in IDD using both in vivo and in vitro approaches.
Materials and methods
Animals
Twenty-four specific pathogen-free (SPF) healthy male Sprague Dawley (SD) rats (8 weeks old; body weight, 200 ±20 g) were purchased from Chengdu Dashuo Biotechnology Co., Ltd. (Chengdu, China). The rats were housed in a pathogen-free environment for 1 week to acclimate to the laboratory conditions, with free access to a standard diet (caloric composition: 70% carbohydrate, 10% fat, and 24% protein; provided by Chengdu Dashuo Biotechnology Co., Ltd.) and water, under a normal 12-h light/dark cycle.
Experimental design
The rats were randomly assigned to 4 groups: the sham group (n = 6), model group (n = 6), shRNA negative control (NC) group (n = 6), and shRNA TRPC3 group (n = 6). An IDD rat model was established using the annulus fibrosus puncture method.31 Twelve hours before model induction, the rats were fasted with ad libitum access to water. The rats were weighed and then intraperitoneally injected with 40 mg/kg of 1% pentobarbital sodium for anesthesia, based on body weight. Once anesthetized, the rats were placed in the supine position with their limbs immobilized. The surgical area was shaved and disinfected with iodine. A longitudinal incision of 3–4 cm was made approx. 0.5 cm to the right of the midline. Each tissue layer was sequentially incised to expose the posterior abdominal wall. The intestinal tract and greater omentum were gently retracted to prevent injury, and the paravertebral lumbar muscles were bluntly dissected. The L4/5 and L5/6 intervertebral discs were exposed, and a 21-gauge needle was used to puncture the annulus fibrosus at 3 points on the right anterior aspect of the vertebral body, directed toward the center of the intervertebral disc. The puncture depth was approx. 2–3 mm, limiting the injury to the full thickness of the annulus fibrosus. Following the injury, the needle was maintained in position for 10 s. After successful puncture, the incision was closed in layers. Rats in the sham surgery group underwent the same skin and tissue incision without intervertebral disc puncture. Postoperatively, routine care was provided, including intramuscular injection of penicillin (8 × 105 U/day, once daily for 3 consecutive days) to prevent infection. The rats were closely monitored postoperatively for food intake, gait, wound infection, and urinary retention.
Three rats were randomly selected from the sham and model groups at 8 weeks after surgery. The rats were anesthetized with isoflurane, followed by cervical dislocation at the atlantoaxial joint. Tissue samples were collected, and the skin, paravertebral muscles, and ligaments were dissected layer by layer. The L5/6 intervertebral disc tissue was rapidly harvested and fixed in 4% paraformaldehyde. To evaluate the success of model establishment and observe morphological changes, hematoxylin and eosin (H&E) staining was performed.
Model establishment was confirmed by pathological examination at 8 weeks after surgery. On the 1st day after successful model establishment, rats in the shRNA NC group received a local intraspinal injection of adeno-associated virus (AAV)-shRNA NC (1 × 1012 μg/kg). Rats in the shRNA TRPC3 group received a local intraspinal injection of AAV-shRNA TRPC3 (1 × 1012 μg/kg). Both AAV-shRNA NC and AAV-shRNA TRPC3 were designed and synthesized by Shanghai Jikai Gene Medical Technology Co., Ltd. (Shanghai, China). After 3 weeks, the rats were anesthetized with an overdose of isoflurane, and NP tissue samples were collected from the intervertebral discs of each group.
H&E staining
Intervertebral disc tissues were first decalcified using a 15% ethylenediaminetetraacetic acid (EDTA) solution. The tissues were then dehydrated and embedded, sectioned at a thickness of 5 μm, stained with hematoxylin for 10–20 min followed by eosin for 3–5 min, and mounted with neutral gum.32 Images were acquired using a Pannoramic 250 FLASH III digital slide scanner (3DHISTECH, Budapest, Hungary).
Immunohistochemistry staining
Intervertebral disc tissues were sectioned, deparaffinized, and rehydrated. The sections were then heated in 10 mM sodium citrate buffer (pH 6.0) at 100°C for 30 min for antigen retrieval and blocked with 3% bovine serum albumin (BSA; cat. No. GC305010; Servicebio, Wuhan, China) for 1 h at room temperature. The sections were subsequently incubated overnight at 4°C with primary antibodies against autophagy-related protein 5 (ATG5; bs-4005R, 1:100; Proteintech, Wuhan, China), Beclin-1 (11306-1-AP, 1:100, Proteintech), and LC3-II (14600-1-AP, 1:200; Proteintech). Afterward, the sections were incubated with a secondary antibody (horseradish peroxidase (HRP)-labeled goat anti-rabbit immunoglobulin G (IgG; H+L) GB23303, 1:100; Servicebio) for 1 h at room temperature. Diaminobenzidine (DAB) color development and hematoxylin counterstaining were then performed.33 Finally, the sections were examined under a light microscope (BA400Digital; Motic China Group Co., Ltd., Xiamen, China), and images were captured using a digital trinocular microscope (BA400 Digital; McAudi, Xiamen, China).
Micro-computed tomography scan
Intervertebral disc tissue samples were scanned using the Micro-CT Scanner software. For three-dimensional analysis, the samples were analyzed using CTAn software (Bruker Micro-CT, Billerica, USA) to obtain the following parameters: structure model index (SMI), trabecular thickness (Tb.Th), bone volume to total volume ratio (BV/TV), trabecular number (Tb.N), and trabecular separation (Tb.Sp).
Primary human NPCs isolation and culture
Nucleus pulposus tissue was obtained during lumbar discectomy, and its morphology was observed under a light microscope (Leica DMI1; Leica Microsystems, Wetzlar, Germany). Following washing with phosphate-buffered saline (PBS), NP tissue samples were digested with 0.25% trypsin and 0.2% collagenase type II at 37°C for 4–6 h. After removal of tissue debris by filtration through a 200-μm filter, purified NPCs were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 supplemented with 10% fetal bovine serum (FBS), 100 μg/mL streptomycin, and 100 μg/mL penicillin at 37°C in a humidified atmosphere containing 5% CO2. Nucleus pulposus cells at passage 2 were used for subsequent in vitro experiments.
Cell transfection and grouping
The cell experiments were divided into 2 parts.
Part I: NPCs were divided into the following groups: control, IL-1β, IL-1β + siRNA NC, IL-1β + siRNA TRPC3, and IL-1β + siRNA TRPC3 + pyrrolidine dithiocarbamate (PDTC; NF-κB inhibitor, 100 μmol/L).
Part II: NPCs were divided into the following groups: control, IL-1β, IL-1β + siRNA NC, IL-1β + siRNA TRPC3, IL-1β + siRNA TRPC3 + 3-methyladenine (3-MA; autophagy inhibitor), and IL-1β + siRNA TRPC3 + rapamycin (RAPA; autophagy activator).
Cell transfection was performed using the RiboFect™ CP Transfection Kit (C10511-05; RiboBio, Guangzhou, China). The lyophilized siRNA was reconstituted in RNase-free water to obtain a 20 μM stock solution. The transfection mixture was prepared by thoroughly mixing 120 μL of RiboFect™ CP Buffer, 12 μL of RiboFect™ CP Reagent, and 10 μL of siRNA, followed by incubation at 37°C. Except for the control group, cells in all other groups were treated with 10 ng/mL IL-1β after cell adherence to induce cellular injury. Simultaneously, cells in the IL-1β + siRNA TRPC3 + 3-MA group were treated with 3-MA (5 mmol/L), and cells in the IL-1β + siRNA TRPC3 + RAPA group were treated with RAPA (250 nmol/L). The corresponding assays were performed 24 h later.
Western blot
Rat intervertebral disc NP tissue or NPCs were lysed for 10 min in radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China). Protein concentrations were quantified using a bicinchoninic acid (BCA) Protein Assay Kit (Beyotime). Protein samples from the supernatant were mixed with an equal volume of sodium dodecyl sulfate (SDS) loading buffer and boiled for 5 min to denature the proteins. Equal amounts of protein were loaded onto a 12% polyacrylamide gel and subjected to electrophoresis for 60–90 min. Subsequently, proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (Merck Millipore, Billerica, USA). To block nonspecific binding, the membranes were incubated with 5% skim milk for 1 h at room temperature. The PVDF membranes were then incubated with primary antibodies overnight at 4°C, followed by incubation with secondary antibodies for 2 h at room temperature.
The antibodies used were as follows: anti-β-actin (cat. No. AC026, 1:50,000), anti-ATG5 (cat. No. A0203, 1:1,000), anti-Beclin-1 (cat. No. A7353, 1:2,000), anti-LC3B (cat. No. A19665, 1:2,000), anti-NF-κB p65 (cat. No. A2547, 1:2,000), anti-phospho-NF-κB p65 (cat. No. AP0123, 1:2,000), and anti-MMP13 (cat. No. A11148, 1:2,000), all purchased from ABclonal Biotechnology Co., Ltd. (Wuhan, China); anti-TRPC3 (cat. No. 77934, 1:1,000) was purchased from Cell Signaling Technology (CST; Danvers, USA); anti-aggrecan (cat. No. DF7561, 1:1,000) and goat anti-rabbit IgG (H+L) HRP (cat. No. S0001, 1:5,000) were purchased from Affinity Biosciences (Beijing, China); and anti-collagen II (cat. No. BS-10589R, 1:2,000) was purchased from Bioss Biotechnology Co., Ltd. (Beijing, China).
Protein bands were visualized using an enhanced chemiluminescence (ECL) kit (Biosharp, Hefei, China), and signals were captured using the Tanon 5200 Multi-System (Tanon, Shanghai, China).
Transmission electron microscope for autophagy observation
A transmission electron microscope (TEM) was used to investigate autophagy in NPCs. First, the NPC samples were subjected to primary fixation with 3% glutaraldehyde to preserve cellular structures. Subsequently, secondary fixation was performed using 1% osmium tetroxide to enhance contrast of the cellular components. Ultrathin sections with a thickness of approximately 60 nm were prepared using an ultramicrotome (Leica Camera AG). To improve electron density and visualization of cellular organelles, the sections were stained with uranyl acetate for 15 min. Subsequently, the sections were briefly stained with lead citrate for 2 min to further enhance contrast. Finally, the stained samples were examined using a JEM-1400 FLASH transmission electron microscope (JEOL, Tokyo, Japan).
Detection of cell apoptosis by flow cytometry
To assess the apoptotic status of NPCs, flow cytometric analysis was performed using the Annexin V–APC/PI Apoptosis Detection Kit (KGA1030; KeyGen Biotech Corp., Ltd., Nanjing, China) according to the manufacturer’s instructions.34 Nucleus pulposus cells were seeded in 6-well plates at a density of 2 × 105 cells per well and incubated at 37°C in a humidified atmosphere containing 5% CO2 to allow cell attachment and growth. Once the cells reached appropriate confluency, apoptosis detection was initiated. A total of 5 μL of Annexin V–FITC and 5 μL of propidium iodide (PI) were added to the cell suspension. The cells were then incubated for 15 min at room temperature in the dark to allow effective binding of the dyes to the cell membranes. After incubation, the stained NPCs were analyzed using a flow cytometer (CytoFLEX; Beckman Coulter, Brea, USA).
Statistical analyses
For statistical analysis, IBM SPSS Statistics v. 25 (IBM Corp., Armonk, USA) was used, and data are presented as the median (minimum–maximum). The Kruskal–Wallis (K–W) test, followed by Dunn’s post hoc test with Bonferroni correction, was applied for multiple-group comparisons. A p < 0.05 was considered statistically significant. Statistical analysis results are presented in Supplementary Tables 1–6.
Results
Inhibition of TRPC3 improved histopathological damage in IDD rats
To investigate the role and mechanisms of TRPC3 in IDD, AAV-shRNA TRPC3 was injected into the intervertebral disc tissue of rats in the animal study. First, intervertebral disc tissues were evaluated using micro-CT, as shown in Table 1. Compared with the sham group, the model group exhibited decreased BV/TV, Tb.N, and Tb.Th values, while Tb.Sp and SMI were increased; however, only BV/TV, Tb.N, and SMI showed statistically significant differences (all p = 0.050). However, no significant difference was observed between the shRNA TRPC3 group and the shRNA NC group (p = 0.127, p = 0.513, p = 0.275). To further evaluate histopathological changes, H&E staining was performed. In the model group, a significant reduction in NPCs was observed compared with the sham group, whereas chondrocytes and the cartilaginous matrix were increased and arranged in clusters. The shRNA NC group did not exhibit notable improvement in pathological conditions compared with the model group. In contrast, the shRNA TRPC3 group exhibited a relatively intact intervertebral disc tissue structure: the annulus fibrosus was arranged in concentric circles with multiple layers of fibrocartilage and showed a denser organization, and the NP was rich in elastic gel-like material, with abundant NPCs and ECM, indicating attenuation of pathological damage (Figure 1). Therefore, these results indicate that TRPC3 knockdown can ameliorate histopathological damage in IDD rats in vivo.
Inhibition of TRPC3 promoted autophagy in the nucleus pulposus of intervertebral discs in vivo
Subsequently, we analyzed the effect of shRNA TRPC3 on autophagy levels in the NP of intervertebral discs in IDD rats using IHC staining. The expression levels of ATG5, Beclin-1, and LC3-II were markedly higher in the model group than in the sham group (all p = 0.050). Moreover, treatment with AAV-shRNA TRPC3 further increased the expression of ATG5, Beclin-1, and LC3-II compared with the shRNA NC group (all p = 0.050) (Figure 2, Supplementary Table 1). In addition, the model group exhibited a marked increase in TRPC3 expression in NP tissue compared with the sham group, as confirmed by western blot analysis (p = 0.05). In contrast, TRPC3 expression levels were significantly decreased in the AAV-shRNA TRPC3-treated group relative to the shRNA NC group (p = 0.024) (Figure 3, Supplementary Table 2). Overall, these findings suggest that high TRPC3 expression may lead to abnormal accumulation of autophagosomes by inhibiting autophagy, thereby aggravating IDD, whereas TRPC3 knockdown may reverse the IDD phenotype by activating protective autophagy signaling.
The knockdown of TRPC3 inhibited the Ca2+/NF-κB pathway to promote autophagy in NPCs in vitro
To further investigate the mechanisms by which TRPC3 regulates autophagy in IDD, in vitro experiments were performed using primary human NPCs. As shown in Figure 4A, intracellular Ca2+ levels were significantly higher in the IL-1β-treated group than in the control group (p = 0.003). Following siRNA-mediated knockdown of TRPC3, Ca2+ levels were significantly reduced compared with those in the shRNA NC group (p = 0.050). In addition, treatment of NPCs with PDTC, a selective NF-κB inhibitor, led to a further reduction in intracellular Ca2+ levels (p = 0.050). Western blot analysis showed that knockdown of TRPC3 attenuated the IL-1β-induced increase in phosphorylated NF-κB p65 (p = 0.011), while total NF-κB p65 expression remained unchanged (p = 0.273) (Figure 4B–D). Notably, PDTC treatment significantly suppressed p-NF-κB p65 phosphorylation in the presence of siRNA TRPC3 (p = 0.050) (Figure 4D, Supplementary Table 3). Furthermore, siRNA-mediated knockdown of TRPC3 increased the expression of ATG5, Beclin-1, and the LC3-II/LC3-I ratio compared with the IL-1β + siRNA NC group (p < 0.05). Moreover, NPCs treated with PDTC in combination with siRNA TRPC3 and IL-1β exhibited further increases in these autophagy-related proteins (all p = 0.050) (Figure 5A–D). In addition, PDTC significantly suppressed the IL-1β-induced increase in TRPC3 expression (p = 0.050) (Figure 5E, Supplementary Table 4). Collectively, these results indicate that TRPC3 activates the NF-κB pathway through Ca2+ influx and promotes autophagy-related protein expression, ultimately exacerbating IDD, whereas combined inhibition of TRPC3 and NF-κB exerts a synergistic effect in enhancing protective autophagy, thereby delaying IDD progression.
The knockdown of TRPC3-activated autophagy promoted apoptosis in NPCs
To further examine the impact of TRPC3-induced autophagy on cell apoptosis, NPCs were treated with rapamycin (RAPA) and 3-methyladenine (3-MA). In the IL-1β group, TEM revealed the presence of autophagosomes and primary lysosomes, whereas the IL-1β + siRNA TRPC3 group exhibited a markedly increased number of autophagosomes and primary lysosomes. Treatment with RAPA further enhanced autophagy, whereas 3-MA reduced autophagic activity (Figure 6). Furthermore, compared with the control group, IL-1β treatment markedly increased NPC apoptosis (p = 0.050). Following siRNA-mediated knockdown of TRPC3, NPC apoptosis was significantly higher than that in the shRNA NC group (p = 0.050). Compared with the IL-1β + siRNA TRPC3 group, treatment with RAPA resulted in a further increase in NPC apoptosis (p = 0.039), whereas 3-MA led to a reduction in apoptosis (Figure 7, Supplementary Table 5).
These results suggest that TRPC3 alleviates IL-1β-induced apoptosis in NPCs by inhibiting autophagy, whereas excessive activation of autophagy exacerbates cell death, indicating a bidirectional role of the TRPC3-autophagy axis in the regulation of apoptosis.
The knockdown of TRPC3 induced autophagy to regulate the expression of IDD-related proteins in NPCs
To investigate the regulatory effect of TRPC3-induced autophagy on ECM degradation in NPCs, western blot analysis was used to detect the expression levels of MMP-13, collagen II, and aggrecan (Figure 8, Supplementary Table 6). Interleukin-1 beta treatment significantly reduced aggrecan (p = 0.007) and collagen II (p = 0.002) expression compared with the control group, while significantly increasing MMP-13 expression (p = 0.002). In addition, knockdown of TRPC3 reversed these expression changes (all p = 0.050). Compared with the IL-1β + siRNA TRPC3 group, treatment with RAPA increased the expression of aggrecan and collagen II and decreased MMP-13 expression (all p = 0.050). In contrast, treatment with 3-MA reduced collagen II expression (p = 0.050) and increased MMP-13 expression (p = 0.050), while no significant difference was observed in aggrecan expression (p = 0.646). Taken together, these results indicate that TRPC3 knockdown reverses IL-1β-induced ECM degradation by promoting autophagy in NPCs.
Discussion
Intervertebral disc degeneration is a leading cause of LBP and neurological compression syndromes. The pathogenesis of IDD is complex and mainly involves excessive mechanical stress, increased apoptosis of NPCs abnormal ECM degradation, dysregulated autophagy, oxidative stress-induced damage, and genetic factors. TRPC3 is a member of the TRP family of cation channels, which are involved in various physiological processes, including calcium homeostasis and cell signaling. This study elucidates a novel role of TRPC3 in IDD pathogenesis, demonstrating that TRPC3 exacerbates disc degeneration by orchestrating Ca2+/NF-κB-mediated suppression of autophagy, thereby providing a new research direction for targeted therapy of IDD.
The intervertebral disc is principally composed of 3 distinct components: the central gelatinous NP, the outer fibrous annulus fibrosus (AF), and the cartilaginous endplate (CEP).35 The NP is predominantly composed of water, proteoglycans, and collagen, along with substantial amounts of elastic proteins, fibronectin, and laminin. In contrast, the AF is primarily composed of collagen fibers rich in type I collagen, whereas the CEP consists of hyaline cartilage.36, 37 In animal models, intervertebral disc tissue in IDD rats exhibits significant structural damage, including a reduction in NPCs, disorganized arrangement of the AF, and abnormal deposition of cartilaginous matrix. Inhibition of TRPC3 ameliorates these pathological changes in rat intervertebral disc tissue. These findings suggest that TRPC3 primarily influences the progression of IDD by regulating cellular functions, such as autophagy and apoptosis, rather than by directly affecting bone structure. Consistent with this, previous studies have shown that members of the TRP channel family, such as TRPV4, regulate ECM homeostasis through ion channel activity in chondrocyte metabolism rather than directly modulating bone remodeling, thereby supporting the conclusions of the present study.38
Autophagy is a crucial cellular process that maintains homeostasis by degrading damaged components through lysosomes and plays a dual role in IDD: moderate autophagy eliminates damaged organelles and protects NPCs, whereas excessive autophagy may induce apoptosis.19 ATG5 is a key regulator of autophagy, facilitating autophagosome formation and promoting autophagic flux.39, 40 The initiation of autophagy largely depends on Beclin-1, and LC3-II is commonly used as a marker of autophagy.41, 42, 43 Our study reveals that under IL-1β stimulation, overexpression of TRPC3 leads to a compensatory upregulation of the autophagy markers ATG5, Beclin-1, and LC3-II, accompanied by ECM degradation, as evidenced by downregulation of collagen II and aggrecan and upregulation of MMP-13. These findings suggest that TRPC3 may cause abnormal accumulation of autophagosomes by blocking autophagic flux, possibly due to impaired lysosomal function. Further experiments confirmed that TRPC3 knockdown or treatment with the autophagy inducer RAPA significantly restored autophagic activity and reversed ECM degradation, whereas the autophagy inhibitor 3-MA exacerbated matrix damage. Together, these results indicate that TRPC3 promotes ECM metabolic imbalance by inhibiting protective autophagy, and that restoration of autophagic flux may be critical for delaying IDD.
Mechanistically, TRPC3 acts as a nonselective cation channel, and its activation leads to intracellular Ca2+ overload.44 Our data demonstrate that IL-1β stimulation significantly elevates intracellular Ca2+ levels in human NPCs, an effect that is effectively mitigated by TRPC3 knockdown. This Ca2+ overload correlates with enhanced phosphorylation of NF-κB p65, suggesting that TRPC3-mediated Ca2+ influx is a critical activator of NF-κB signaling. In addition, Ca2+ may activate the IκB kinase (IKK) complex via calcium/calmodulin-dependent protein kinase II (CaMKII), thereby promoting IκBα degradation and subsequent nuclear translocation of NF-κB.45 Notably, the NF-κB inhibitor PDTC not only suppressed p-NF-κB p65 phosphorylation but also attenuated IL-1β-induced TRPC3 upregulation, revealing a self-reinforcing “TRPC3–Ca2+–NF-κB–TRPC3” positive feedback loop. This vicious cycle amplifies apoptotic responses and ECM degradation, thereby promoting IDD progression. Knockdown of TRPC3 or treatment with the NF-κB inhibitor PDTC reversed this phenomenon and synergistically enhanced the expression of ATG5, Beclin-1, and LC3-II. These findings suggest that TRPC3 negatively regulates autophagy through the Ca2+/NF-κB axis, and that the underlying mechanism may involve NF-κB-mediated transcriptional repression of autophagy-related genes, such as the ATG5 promoter.46
One of the characteristic features of IDD is apoptosis of NPCs.47 The interplay between autophagy and apoptosis has attracted considerable attention in IDD research. In this study, we observed that IL-1β-induced apoptosis in NPCs was further exacerbated by TRPC3 knockdown. In addition, the autophagy activator RAPA enhanced apoptosis, whereas the autophagy inhibitor 3-MA attenuated apoptotic responses. These findings suggest bidirectional regulation of autophagy at different stages: moderate autophagy inhibits apoptosis by clearing damaged mitochondria, whereas excessive autophagy – such as “autophagic stress” caused by impaired autophagic flux – may promote apoptosis by activating caspase pathways or depleting pro-survival signals.48 TRPC3 knockdown may trigger this latter effect by inducing excessive autophagy, including accumulation of autophagosomes.
Extracellular matrix degradation is a central feature of IDD,49 primarily driven by an imbalance among MMPs, aggrecan, and collagen II. During the early stages of IDD, there is a notable increase in the production of type II collagen in the NP, suggesting an attempt by the tissue to initiate repair mechanisms. As degeneration progresses, type II collagen content markedly decreases, while type I collagen forms more pronounced fibrous structures in the AF and NP.50 In addition, a key hallmark of degeneration is alteration of proteoglycans, particularly degradation of aggrecan, a major polymeric proteoglycan of the intervertebral disc.51 Aggrecan, with its glycosaminoglycan side chains containing numerous negatively charged groups, confers unique permeability to the NP, allowing it to maintain a swollen state under compressive loads.52 Moreover, recent studies have demonstrated that ECM degradation is an early and critical event in IDD, involving metalloproteinases such as MMP-1, MMP-3, and MMP-13, which play key roles and are considered risk genes associated with disc degeneration.53, 54 Our study demonstrates that TRPC3 knockdown reverses IL-1β-induced upregulation of MMP-13 and downregulation of collagen II and aggrecan by activating autophagy. Mechanistically, autophagy may maintain ECM homeostasis through several pathways: 1) degrading damaged MMP precursors55; 2) inhibiting NF-κB-mediated MMP-13 transcription via the mTORC1 pathway56; and 3) enhancing the anabolic function of NPCs to promote secretion of ECM components.57 Notably, the autophagy inhibitor 3-MA partially reverses the protective effects of TRPC3 knockdown on the ECM, further supporting the central role of autophagy. In addition, NF-κB, as a key transcription factor regulating MMP-13 expression, may directly reduce ECM degradation when inhibited, while TRPC3 knockdown exerts a synergistic protective effect through a dual mechanism involving autophagy activation and NF-κB inhibition.
Limitations of the study
This study has several limitations. First, we only conducted an initial exploration of the role of TRPC3 in IDD. In addition, the rat IDD model cannot fully recapitulate the chronic degenerative process observed in humans; therefore, the effects and underlying mechanisms of TRPC3 on ECM metabolism in IDD in vivo require further investigation. Although this study elucidates the mechanism by which TRPC3 regulates autophagy through the Ca2+/NF-κB pathway, the pathophysiology of IDD is complex and multifactorial, potentially involving multiple signaling pathways and molecular mechanisms beyond those examined here. Future research should explore additional signaling pathways and molecular mechanisms involved in IDD and their relationships with TRPC3. In addition, the potential involvement of other Ca2+ channels, such as TRPV4, cannot be excluded and warrants further validation of the functional specificity of TRPC3 using gene-editing approaches, such as CRISPR–Cas9.
Conclusions
The results of this study indicate that TRPC3 promotes autophagy by regulating the Ca2+/NF-κB pathway, induces apoptosis of NPCs and ECM degradation, and thereby contributes to the development of IDD. These findings highlight the importance of TRPC3 as a key therapeutic target in IDD and provide potential strategies for the treatment of this condition.
Future studies may focus on the development of small-molecule drugs targeting TRPC3, particularly highly selective TRPC3 inhibitors, as well as the application of nanodelivery technologies to enhance the clinical translation potential of TRPC3-targeted therapies for IDD. In addition, exploring combination strategies involving TRPC3 inhibitors together with NF-κB antagonists or autophagy modulators may synergistically enhance the suppression of IDD progression.
Supplementary files
The supplementary materials are available at https://doi.org/10.5281/zenodo.16352711. The package contains the following files:
Supplementary Table 1. Kruskal–Wallis test results for Figure 2.
Supplementary Table 2. Kruskal–Wallis test results for Figure 3.
Supplementary Table 3. Kruskal–Wallis test results for Figure 4.
Supplementary Table 4. Kruskal–Wallis test results for Figure 5.
Supplementary Table 5. Kruskal–Wallis test results for Figure 7.
Supplementary Table 6. Kruskal–Wallis test results for Figure 8.
Data Availability Statement
The raw data of the current study are openly available in Zenodo repository at https://doi.org/10.5281/zenodo.16352556.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)