Abstract
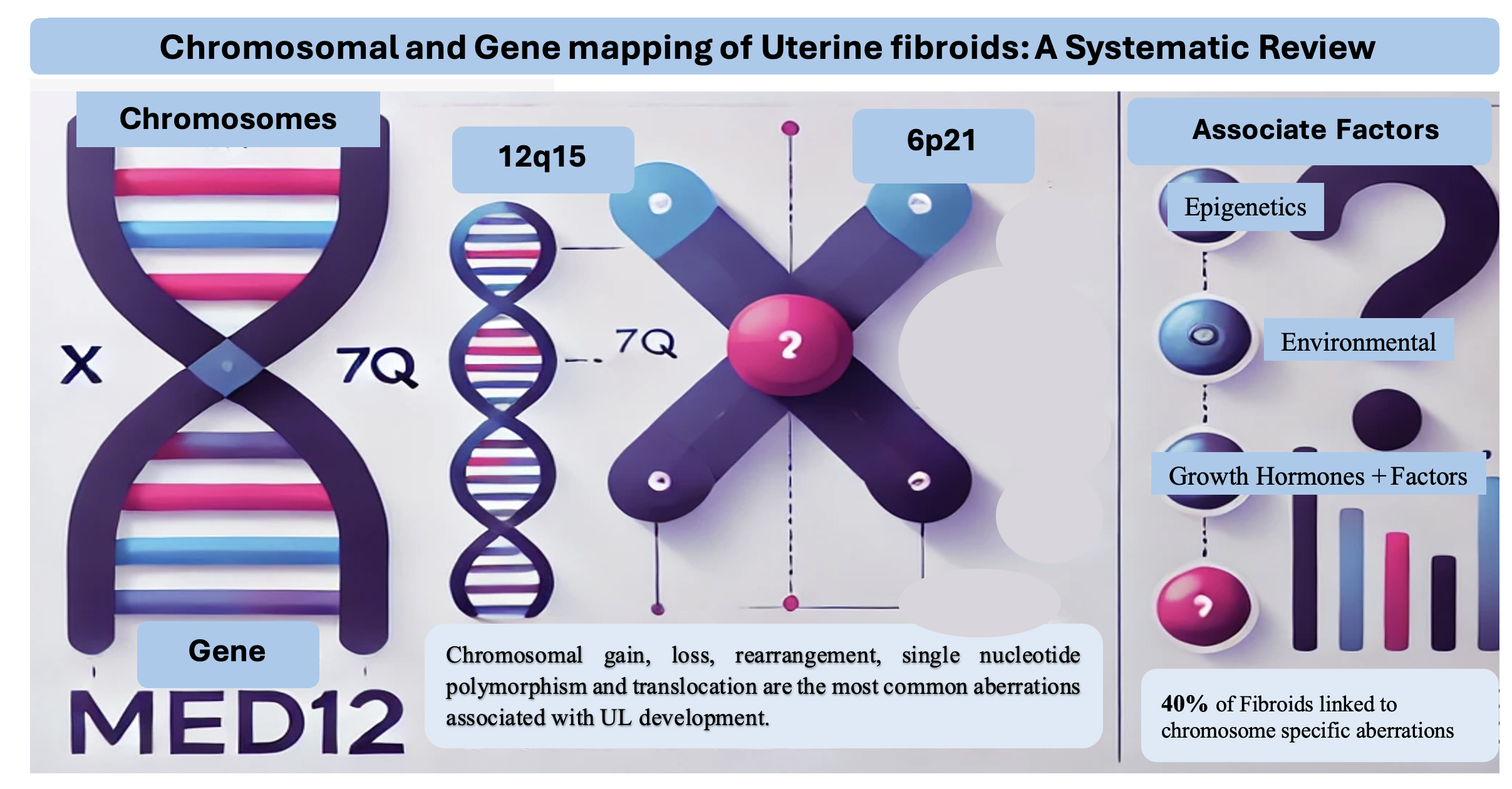
Genetic mutations and their phenotypic manifestation have been recognized as critical factors in tumorigenesis. However, the relationship between these mutations and the pathogenesis of uterine leiomyomas (UL) remains inadequately characterized. There is compelling evidence to suggest a genetic underpinning in UL development, alongside influences from epigenetics, environmental stimuli, growth hormones, and growth factors. A plethora of studies have tried to elucidate the genetic and epigenetic etiologies associated with UL, but the definitive implications of these findings remain unclear. An extensive systematic review was conducted to investigate the genetic etiologies of UL. This systematic review aimed to consolidate current knowledge on genetic and epigenetic causes of UL, offering a comprehensive perspective on the evidence and its relevance in other solid tumors. A secondary focus was to identify the most significant genetic association with the genesis of UL. A total of 60 articles were identified, and 10 chromosomes and 51 genes were found to be implicated in the development of UL. The main trend in fibroid research focuses on genetic abnormalities and aberrations as the etiology of UL development. It has been estimated that 40% of UL can be associated with chromosome-specific aberrations. Chromosomal gain, loss, rearrangement, single nucleotide polymorphism (SNP), and translocation are the most common aberrations associated with UL development. The most recurrent ones include chromosome X and 7q deletions, and rearrangements of 12q15, 6p21 and 10q22. MED12 has been identified as a gene of particular importance in the development of UL.
Key words: uterine fibroids, tumorigenesis, genotyping, leiomyomas, myoma genetics
Introduction
Ontogenetically, the uterus is divided into 2 distinct organs: the inner archimetra and the outer neometra. The archimetra encompasses the endometrium and the subendometrial layer of the myometrium (stratum subvasculare). It is of paramesonephric origin, making it the oldest part of the uterus. The neometra includes the stratum vasculare and the stratum supravasculare of the myometrium, constituting the outer main bulk of the muscular wall.1
From an immunocytochemical perspective, the subendometrial layer of the myometrium (stratum vasculare) exhibits a cyclical pattern of estrogen and progesterone (PR) receptor expression, similar to that of the endometrium. In contrast, the outer neometra does not display this cyclical receptor expression. This finding correlates with the functionality of both ‘organs,’ as the endometrial-subendometrial unit is involved in the cyclical preparation for implantation, while the outer bulk of the uterus requires continuous growth for parturition, necessitating constant exposure to estrogen and PR.2
Uterine leiomyomas (UL), also known as uterine fibroids or myomas, are noncancerous growths of the myometrial layer of the uterus. They occur in up to 70% of women, making them the most common reason for gynecologic surgery referrals. Although no single etiology has been identified to directly mediate the development of UL, a multifactorial approach has been adopted to study the causes of these benign masses, with genetics, hormones and growth factors believed to interact in their pathophysiology.3
Various attempts to isolate the aberrant genetic etiology of UL have been reported in the literature, leading to numerous associations between chromosomes, genes and their development.
Objectives
The primary objective of this article was to provide a concise and comprehensive review of the key genetic etiologies involved in the development of UL. The secondary objective was to explore how aberrations in specific chromosome regions might lead to particular pathologies.
Methods
Inclusion criteria
Articles were screened based on the following criteria: level of evidence (1–4), focus on leiomyoma, uterine fibroid, or uterine myoma and their genetics, etiology or genomic analysis, and publication in English. Articles were critically analyzed for their reporting of clinical or functional outcomes, as well as clinical follow-up if available. Animal studies, pilot studies and studies with no clear correlation or specific patient outcomes were excluded. Articles related to benign smooth muscle tumors and gastrointestinal stromal tumors (GIST) were also reviewed.
Search strategy
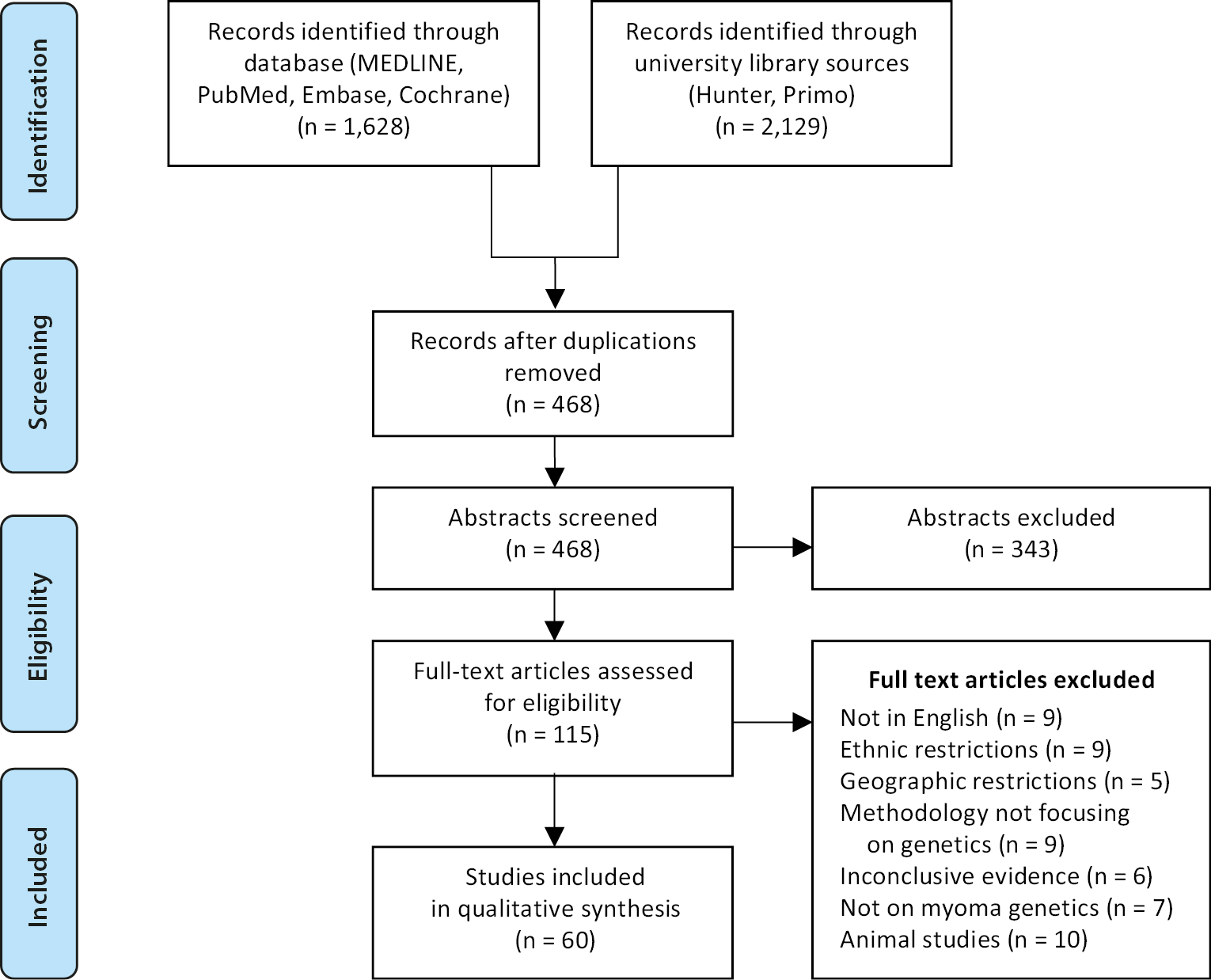
A comprehensive search for scientific papers reporting genetic etiologies for UL was conducted using the following databases: MEDLINE, PubMed, Embase, Cochrane Library, the American Association for Cancer Research Publications, and the Hunter and Primo search engines of the University of Saint George's (London, UK) and the University of Aberdeen (Aberdeen, UK), respectively. The final list of articles was screened to identify any additional potential sources. Figure 1 illustrates our search strategy as well as the inclusion and exclusion criteria. Our study included all genes found to be associated with UL, and 6 papers focusing on benign smooth muscle tumors were also included. The details of the genes presented in Table 1, Table 2 were retrieved from a total of 60 papers.
Data analysis
The selected articles were methodically assessed for validity and bias using the Critical Appraisal Skills Program (CASP) to enhance the rigor of this review. The following domains were evaluated to determine whether the criteria were “clearly met”, “clearly not met” or “cannot tell” if the criteria were met, where the last option was used for cases the authors could not assess. Randomized controlled trials (RCTs) were appraised using the modified Cochrane scale. This review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.
Results
In total, 10 chromosomes and 51 genes were involved in UL (Table 1). Five types of aberrations were identified in 49 of these genes: chromosomal gain, chromosomal loss, rearrangement, single nucleotide polymorphism (SNP), and translocation (Table 2), with chromosomal gain being the most common, appearing in 31% of cases. Although sample frequency could not be compared across studies, the following paragraphs detail the chromosomes and genes most strongly associated with UL development (Supplementary Fig. 1).
Chromosome 1
Seven genes identified on chromosome 1 have been found to contribute to UL pathogenesis through diverse mechanisms of alteration. Genes involved on the “p”-arm of chromosome 1 are usually involved in transcription regulation. On the “p”-arm of the chromosome, JUN, AJAP and NPHP-4 are located on p21.3-p31.3 and p36 breakpoints, respectively.4 Genes AJAP and NPHP-4 are usually the translocation partners of NCOA1 on chromosome 2 (P-24), which is the translocation found to be occurring in 1–3% of UL cases.4 Additionally, deletions of 1p are observed frequently in UL, and they tend to be associated with distinct histopathological features and possible malignant progression. The gene KIF1B is most likely affected with 1p deletions observed in UL.5
On the “q”-arm of the chromosome, the genes CSK1 and FH are located at q22 and q43, respectively. The FH gene is a tumor suppressor gene (TSG) involved in DNA double-strand break repair,5 while CSK1 is a cell cycle regulator. Alterations in the FH gene in UL can result from chromosomal loss or a missense mutation,6 while aberrations in CSK1 are due to chromosomal gain.7 Lastly, the 1p13 region of chromosome 1 is subject to epigenetic regulation through hypermethylation in UL and contains the GSTM5 gene.8
Chromosome 2
On chromosome 2, certain areas have been identified to exhibit epigenetic changes, including KLF11, a TSG located on 2p25. KLF11 is a target of PR and antiprogesterone agents in UL tissues. It is downregulated in UL by repressing most endometrial cytochrome enzymes in Ishikawa cells. KLF11 binds to the guanine (G) and cytosine (C) DNA sequence (GC) elements of the estrogen-metabolizing enzyme CYP3A4 promoter, repressing its promoter and enzymatic function. This leads to selective deacetylation of the CYP3A4 promoter. KLF11 recruits the coreceptor SIN3A/HDAC, and HDAC pharmacologically reverses this inhibition.8
Furthermore, 2p13, which contains the genes TET1 and TET3, undergoes epigenetic changes in UL. These genes code for an enzyme that regulates DNA methylation. An increase in 5-hmC levels is associated with the upregulation of TET1 or TET3, leading to transient changes in DNA methylation. This alteration affects the cellular environment and promotes increased proliferation. The suspected association of 2q22.1 with increased tumor size, however, is not linked to the risk of UL.5
Chromosome 3
Insertions, translocations and deletions involving chromosome 3 – such as t(3;7)(p11;p11), ins(2;3)(q31;p12p25) and del(3)(p24) – have been documented in UL, although they are less common compared to other chromosomal abnormalities.9 Certain regions of the short arm (“p” arm), particularly 3p22, are also subject to epigenetic modifications. This locus includes the DLEC1 gene, a known TSG. Hypermethylation of DLEC1 has been implicated in tumorigenesis and has been reported in both esophageal cancers and UL.8 Region 3p22 and its adjacent loci are susceptible to various pathogenic mechanisms, including structural alterations and epigenetic silencing.
Chromosomes 4 and 5
The involvement of growth factors in UL has been mapped to chromosomes 4 and 5. On chromosome 4, EGF and bFGF are located at q25 and q28, respectively. On chromosome 5, aFGF and HBEGF are both situated at 5q31.10 However, to date, no dedicated studies have specifically investigated the frequency or expression levels of aFGF and HBEGF in this chromosomal region in the context of UL.
EGF, aFGF and bFGF not only share a similar protein size of approx. 100,000–105,000 Da but also exhibit increased expression levels in UL. In contrast, HBEGF expression appears to be decreased. Additionally, both aFGF and bFGF play key roles in promoting angiogenesis, while EGF and HBEGF primarily contribute to cell proliferation.11
The 4q34 region is a site of epigenetic alteration and is associated with the MORF gene, which is disrupted by the t(10;17) translocation. This disruption results in a distinct gene expression pattern that is implicated in the pathogenesis of UL.8
Chromosome 6
Chromosome 6 harbors the HMGA1 and ESR1 genes, located at positions p22 and q25.1, respectively. Alterations in HMGA1 often involve gene rearrangements that can lead to its overexpression. Such aberrations have been identified in approx. 3% of UL cases, suggesting a potential, though limited, role in the pathogenesis of these tumors.5
However, HMGA1 aberration are more commonly secondary changes, with 45-fold increased expression in sample studies.12 HMGA1 is important in gene transcription regulation and nucleosome phasing as it regulates genes close or within A+T rich regions.10 HMGA1 was also found to be altered in other benign mesenchymal tumors such as lipomas and hamartomas.5
Estrogen and PR receptors in UL exhibit cyclical changes similar to those observed in normal uterine tissues during the menstrual cycle. Estrogen levels decrease in the postovulatory phase but remain consistently higher in submucosal compared to subserosal UL, in both the proliferative and secretory phases, paralleling PR levels.13 In leiomyoma smooth muscle cells, estrogens exert their effects through both estrogen receptor alpha (ER-α) and beta (ER-β). However, in endothelial and connective tissues, estrogen acts primarily via ER-β. Estrogen receptor alpha is expressed exclusively in smooth muscle cells, with its distribution varying across different UL subtypes at the subcellular level. In contrast, ER-β is widely distributed across smooth muscle, endothelial and connective tissue cells and is consistently localized within the nucleus. Notably, subcellular variations of ER-β expression were observed only in smooth muscle cells.14
Estrogen receptor alpha is present in nucleus and cytoplasm, while ER-β is localized only in nuclei. Estrogen receptor alpha and ER-β proto-oncogenes expressions do not differ between UL and normal myometrium, but in both cases ER-α expression is always higher than ER-β expression. Estrogen receptor alpha expression is also higher in both when compared to normal endometrium.15
Estrogen receptor alpha mRNA and protein levels in multiple ULs were significantly lower than those in solitary ULs. ER-β mRNA and protein levels were significantly higher in multiple ULs than in solitary ones, irrespective of stage of menstrual cycle. In both, ER-a expression was higher than that of ER-β. Estrogen receptor alpha concentration was also correlated to E2 concentration in multiple and solitary ULs. Difference in ER-α and ER-β expression between multiple and solitary ULs may be responsible for course of disease subtypes.16 Expression of IGF-1 mRNA also increases in UL and has been correlated to the upregulation of estrogen by ER-1. The involvement of ER-β is also probable but has not yet been found to be statistically significant.15
Estrogen receptor alpha to ER-β expression ratio rather than individual expression levels can also determine fraction of DNA binding homodimers of ER-α, and possibly the growth potential of myoma.17, 18
ERs are also found to be changed epigenetically in ULs in different ways. Normally, hypermethylation of the protooncogene at the site 6p21 levels are inversely related to ER expression levels. Hypomethylation to oncogene leads to development of tumorigenesis and ER-α gene, which is highly expressed in UL. Seven CpG sites in distal promoter region of ER-α gene variation were seen compared to normal myometrium. Hypomethylation at this site is correlated with increased ER expression. In addition, HDAC6 was found to be strongly stained in UL's cytoplasm. Silencing of HDAC6 by siRNA led to decreased ER-α protein levels but not ER-α mRNA, suggesting that it promoted degradation of ER-α protein in lysosome.8
The enzyme 17β-hydroxysteroid dehydrogenase is also altered by polymorphisms in UL, leading to reduced expression. As a result, its substrate, estradiol, accumulates and binds more strongly to ER than its product, estrone.17 Activation of estradiol further stimulates the pro-mitogenic PI3K/mTOR signaling pathway.19, 20
Chromosome 7
Chromosome 7 is implicated in the development of UL, with 5 genes undergoing chromosomal loss: CUX1, ORC5L, LHFPL3, ZNHIT1, and CUL1. The most recurrent aberration among these involves the CUX1 gene at locus 7q22.1, with an overall frequency of 14% in all UL.21
CUX1 has a tumor suppressor gene function.20, 21 In UL, CUX1 chromosomal loss induces a signaling pathway that results in elevated tumor growth and proneness to PI3K-AKT repression through transcriptionally downregulating PIK3IP1 (PI3K inhibitor). Similarly, CUX1 is a compelling candidate for the tumor suppressor gene. Its chromosomal loss has been implicated in the development of leukemia and myeloproliferative disorders, where it demonstrated around 45.8% reduction in the expression level.20 The prevalence of CUX1 chromosomal loss in hematologic malignancies has been estimated to be 15–25% of acute myeloid leukemias and myelodysplastic syndrome (MDP),21 and 50% of therapy-related myeloid neoplasms.20
On the other hand, CUX1 can act as a proto-oncogene and contribute to tumor progression20, 21 by resisting apoptosis. It is also involved in the enzyme reaction of the kinases ataxia-telangiectasia mutated (ATM) and ATR (ATM- and Rad3-related) in response to DNA damage.21
For instance, a study conducted by Ramdzan et al.19 showed that 71% of 885 human tumors expressed CUX1 chromosomal gain. Hence, both decreased and increased expression of CUX1 can contribute to tumorigenesis by functioning either as a tumor suppressor gene or as oncogene.21 Finally, CUX1 was shown to be involved in an inversion-induced fusion chromosomal rearrangement (CUX1-AGR3, 7q22-p21) in 1 of the samples.24
Located on the same chromosomal locus as CUX1 at 7q22.1, the ZNHIT1 gene is found to contribute to UL development in 1 out of 38 samples.3 The other genes on chromosome 7 that are involved in UL include ORC5L on 7q22,25 LHFPL3 on 7q22.2-q22.3 (3 out of 4 samples)26 and CUL1 on 7q31 (4 out of 38 samples).3 Nonetheless, it is important to mention that the sample number for the genes ZNHIT1, LHFPL3, CUL1, and ORC5L were statistically insignificant and hence their involvement in the pathogenesis of UL cannot be confirmed.
Moreover, the other 2 genes on chromosome 7 are both growth factors and include PDGF1 on 7p22, and ACTIVIN-A on 7p14.33
Chromosome 9
Mehine et al. observed a total of 2 genes on chromosome 9 in 2 out of 6 samples.31 One such gene is CDKN2A, a tumor suppressor located on chromosome 9p21.3, which undergoes chromosomal loss and produces 2 distinct proteins through alternative splicing.7 The 2nd one is concerning a chromosomal loss in ENG on 9q33.1-34.2.7 Epigenetically, the 9q21 region, which contains the DAPK1 gene, undergoes hypermethylation. DAPK1, a TSG, is frequently altered in various human cancers due to its location within a promoter region.8 Epigenetic changes and chromosomal aberrations at 9p21 and 9p21.3 occur at closely located loci. Further studies are needed to determine whether a correlation exists between them. However, there is no large study sample on chromosome 9 to suggest its significance in UL.
Chromosome 10 and 11
The KAT6B (also known as MORF) gene is located on chromosome 10q22. It encodes a histone acetyltransferase (an enzyme that reduces the affinity of nucleosomes for DNA), thereby enhancing DNA accessibility to transcription factors, i.e., a crucial mechanism in epigenetic regulation. Its most frequent translocation partner is 17q21, a region linked to 17β-hydroxysteroid dehydrogenase. However, the significance of this translocation appears limited, as it has been observed in only 9 out of 171 samples. This suggests it may not be a major event in the studied context.32, 34, 35, 36
Other studied alterations at chromosome 10 are SNP alterations on 10q24.33; the alterations also occur at 11q15.5. 10q24.33 is the location for SLK and OBFC1, and the 11p15.5 includes BET1L, RIC8A and SIRT3. Most of the genes involved are TSGs and they were observed only in a small sample of patients.37 The site 10q 25 is found to be altered epigenetically with miR-21. The site contains the PDCD-4 gene, a tumor suppressor that has been found to be downregulated in various types of cancer.8
Chromosome 12
A total of 4 genes on chromosome 12 are involved in UL through chromosomal gain. They include KRAS,7 ELK3,7 HMGA2,28 and IGF-1.33 KRAS is located on 12p11.22-p13.3 and is a proto-oncogene whose chromosomal gain also leads to non-small cell lung cancer in 30% of the 1.6 million per year lung cancer cases.34 The other 2 genes (EKL3 on 12q21.2-q24.37 and HMGA2 on 12q14-15)28 are both involved in embryogenic development. However, the data samples pertaining to both KRAS and EKL3 are only 3/6 samples,7 and hence not proven to be significant. Nonetheless, in a study HMGA2 chromosomal gain was reported in 68/180 samples,35 which results in upregulation of IGF2BP2.23 The last gene on chromosome 12 to be involved in UL is the growth factor IGF-1 located on 12q23. This also leads to activation of IGF-2, along with IGF-1.33 Moreover, larger fibroids were shown to have elevated IGF-1 levels in comparison to smaller ones.33 It is observed from our data that the affected locus of IGF-1 is within that of ELK3, which might encourage more investigations regarding correlation between the 2 genes.
Epigenetics change of the HMGA2 gene in UL has also been reported. The gene contains structural DNA-binding domains and acts as transcriptional regulation. let-7 expression is upregulated in UL. It targets HMGA2, which is implicated in pathogenesis of mesenchymal tumors like leiomyoma, lipoma and hamartomas.8
The dysregulation of IGF-1 is significantly deregulated in UL. IGFBP2 is the 2nd most expressed gene in the HMGA2 subtype of UL, as HMGA2 binds to its AT rich domain and regulates its expression. Subsequently IGF2BP2 induces the expression of IGF mRNA; IGF was detected to be upregulated in the majority of HMGA2 subtypes. IGF2 is then able to bind to IGF1 receptor and promote growth. Also, HMGA2, IGFBP2 and IGF2 are all not expressed in adult tissue, which may indicate they are functionally related. However, ADAM12 has to be the most uniquely upregulated gene in the MED 12 subtype, and PAPPA2 is one among the highest expressed genes in HMGA2 subtypes. Both genes increase the expression of IGFBP, which inhibits the IGF1 receptor action and inhibits IGF-1 role in migration and proliferation in smooth muscle. Therefore, the action of IGFBP5 is still not well understood as to whether it induces or decreases cancer development.5
Two genes on chromosome 12 were also relevant to UL genesis, which are MED12 on Xq13 and COL4A5/6 on Xq22.3. Chromosomal loss of COL4A5/6 was observed on 4/38 samples of UL,21 as well as in Alport syndrome.20 MED 12 mutations represent one of the most frequent genetic aberrations to be discovered, accounting to 70% of UL.36 Along with HMGA 1&2, they represent around 80–90%.6
Moreover, the mutual exclusivity of MED12, HMGA2, FH, and COLA4A5/6 genetic aberrations was reported.6 Nonetheless, amongst these cases, up to 60% coexisted with different genetic alterations, including CUX1 deletions and HMGA1 mutation.6 MED12 mutations also exhibited an increased expression of the gene RAD51B.6 Interestingly, 10–20% of leiomyosarcoma were observed to contain MED 12 mutations.20 The malfunction resulting from mutated MED12 is believed to be caused by an absent transcriptional inhibition affecting cell cycle regulators such as Cdk8 and cyclin C, as well as the WNT signaling pathway.6 MED12 engages with REST and β-catenin, and both participate in the WNT pathway.20 This can eventually lead to a decreased expression of REST, entailing the upregulation of mTORC.20
Chromosome 14
On chromosome 14, 3 important genes are implicated in UL: ESR2, TGF-β and RAD51B. While ESR2 (also referred to as ESR-B) has already been discussed alongside ESR1 (ESR-A), it is worth noting that ESR2 is located near TGF-β and RAD51B at chromosomal regions 14q22, 14q23–24 and 14q24, respectively.36 This may require further investigation regarding their association and the frequency of these genetic aberrations occurring together in UL.
In ULs, there is also increased expression of TGF-β; however, its signaling mechanism remains poorly understood. TGFB1 is linked to increased expression of fibromodulin, which might lead to fibrotic properties of UL. The expression of TGFB3 increases the manifestation of extracellular matrix (ECM) components such as COL1A1 and FN1, while decreasing the expression of genes associated with ECM degradation.37
RAD51B is the preferred translocation partner of HMGA2, and this translocation is associated with decreased HMGA2 expression. However, HMGA1 and HMGA2 rearrangements frequently target other regions in other mesenchymal tumors. Biallelic loss of RAD51B was observed in ULs without the involvement of HMGA2. The gene encodes a double strand break repair enzyme and may play a role in promoting genomic instability. It is also observed to be uniquely overexpressed in ULs with MED12 subtypes. However, whether increased expression of this gene is involved in UL development and growth requires further study.38
Chromosome 17
Two genes were reported on chromosome 17. The first one is the TSG TP53 (located on 17p13.2) that demonstrated chromosomal loss only in 2 out of 6 samples.7 The 2nd genetic aberration is a single nucleotide polymorphism 17 beta-hydroxysteroid, located on 17q21.17 The site 17q21 is also a site of hypermethylation in ULs. The epigenetic change may involve KRT19, another TSG. Hypermethylation can lead to inhibition of the TSG, resulting in tumorigenesis.8
Chromosome 22
The literature identifies a total of 4 genes located on chromosome 22; PDFGB on 22q13.1-22q13.37 and TNRC6B on 22q13.1.39 PDFGB leads to UL development through genetic chromosomal loss and has been reported in 4 out of 6 samples.7 PDGFB mutation can also lead to ovarian cancer.10 DEPDC5 is located on chromosome 22q12.2–q12.3 and is implicated in hepatocellular carcinoma.40 SMARCB1, located at 22q11, undergoes rearrangements that can lead to biallelic loss of function, resulting in the inactivation of this TSG.23
Chromosome X
Both the “p”- and “q”-arms of the X chromosome are susceptible to epigenetic modifications, particularly at loci Xp11 and Xq26. In both regions, the predominant alteration observed is DNA hypomethylation. The gene located at Xp11 encodes a cell cycle progression inhibitor, and its reduced expression due to hypomethylation may contribute to tumorigenesis.8
Chromosome 1
The FH gene on chromosome 1 is strongly associated with HLRCC. However, alterations are observed in only a small subset of UL cases, making its prevalence difficult to quantify.6 Additional pathologies and malignancies have been related to the CSK1 and FH genes.26 For instance, while CSK-1 is implicated in breast, prostate, esophagus, and bladder cancers,27 FH is involved in HLRCC and uterine leiomyosarcoma.6 We examined JUN and CSK-1 genes in 6 samples, with JUN showing up in 1 and CSK-1 in 2 of them.7 However, a study by Lehtonen et al. reported FH aberrations in 5 out of 153 samples.33
Similarly, epigenetic regulation also affects some regions of chromosome 1. Uterine leiomyomas involving the GSTM5 gene are susceptible to hypermethylation at the 1p13 region of chromosome 1. Hypermethylation of this region results in reduced gene expressions, which raises the risk of cancer and exposure to carcinogens.8 GSTM5 gene encodes an enzyme involved in the detoxification of electrophilic compounds, including carcinogens.
Chromosome 2
Regions of chromosome 2 have also been shown to undergo epigenetic alterations. In UL tissues, expression of KLF11 is reduced, leading to the suppression of several endometrial enzymes.8 The role of HDAC was implicated through KLF11’s repression of the CYP3A4 promoter, and its recruitment of the corepressor complex SIN3A/HDAC to mediate this effect.8 The TET1/TET3 genes on 2p13 control DNA methylation. An increase in DNA methylation, driven by elevated 5-hmC levels, can influence the cellular environment by promoting cell proliferation. However, the risk of developing UL is not associated with the 2q22.1 region.8
Chromosome 3, 4 and 5
Chromosome 3 insertions, translocations and deletions are consistent but less prevalent in uterine fibroids. 3p22 is the region most frequently affected by epigenetic modification.9 Hypermethylation of this site contributes to tumorigenesis in UL and esophageal cancers. While HBEGF appears to have reduced expression of EGF, both aFGF and bFGF exhibit comparable protein sizes. Both genes contribute to angiogenesis and cell proliferation, and a mutation in the MORF gene at 4q34 is associated with a distinct expression pattern involved in the etiology of UL.8
Chromosome 6
HMGA-1 and ER-1 genes are found on chromosome 6. The nucleosome-phasing and gene transcription regulatory protein HMGA1 has been reported to be altered in approx. 3% of uterine fibroids.11 Both UL and normal myometrium expressed the ER-α and ER-β proto-oncogenes, with ER-α levels being higher than those of ER-β. However, multiple UL exhibited significantly lower levels of ER-α mRNA and protein compared to single UL. Regardless of the menstrual cycle phase, multiple ULs consistently exhibited significantly higher levels of ER-β mRNA and protein compared to solitary ULs.
IGF-1 mRNA expression is also elevated in UL and has been linked to the increase in estrogen induced by ER-1. Beyond individual expression levels, the ratio of ER-α to ER-β may also predict the proportion of ER-DNA binding as homodimers and, potentially, the myoma’s tendency to grow. Hypermethylation of proto-oncogenes can also lead to epigenetic alterations in ERs. Alterations in the ER-α gene have been observed during tumorigenesis, primarily due to hypomethylation of the oncogene when compared to normal myometrium. Additionally, polymorphisms in the 17β-hydroxysteroid dehydrogenase enzyme reduce its expression. This reduction, combined with estradiol activation, can further stimulate the pro-mitogenic PI3K/mTOR signaling pathway.11
Chromosome 7
Seven genes located on chromosome 7 have been associated with the development of UL, with CUX1 being the most frequently affected. CUX1 functions primarily as a TSG, and its activation has been shown to inhibit the PI3K-AKT signaling pathway, thereby contributing to tumor progression. Interestingly, CUX1 also exhibits characteristics of a proto-oncogene, as it can promote tumorigenesis by resisting apoptosis. In addition to its roles in UL, CUX1 has been implicated in leukemia and myeloproliferative disorders.20, 21 Moreover, it is involved in regulating the enzymatic activity of ATM and ATR kinases, which are critical for the cellular response to DNA damage. In 1 out of 38 UL samples, ZNHIT1 and CUL1 were also identified at the same chromosomal locus as CUX1. Notably, CUX1 was found to participate in a chromosomal rearrangement involving an inversion-induced gene fusion.14
Chromosome 9
Two genes located on chromosome 9 – CDKN2A (a tumor suppressor gene) and ENG – were identified in 2 out of 6 analyzed samples. ENG exhibited a chromosomal deletion. Notably, chromosomal abnormalities at loci 9p21 and 9p21.3 occur in proximity, while the DAPK1 gene, situated at 9q21, is known to be epigenetically susceptible to hypermethylation. Despite these findings, the role of chromosome 9 in UL pathogenesis has not yet been thoroughly investigated in large-scale studies.8
Chromosome 10
KAT6B, located on chromosome 10, encodes a histone acetyltransferase that plays a critical role in epigenetic regulation. Several SNPs, including BET1L, RIC8A and SIRT3, have been identified with alterations on 10q24.33 and 11p15.5. Additionally, 10q25 undergoes epigenetic modification involving miR-21. This is associated with the downregulation of PDCD4, a gene observed to be suppressed in various malignancies.16
Chromosome 12
Four genes (KRAS, ELK3, HMGA2, and IGF-1) have been implicated in UL through chromosomal gain on chromosome 12. KRAS is a proto-oncogene known to contribute to non-small cell lung cancer, while HMGA2 and ELK3 are involved in embryonic development. Chromosomal gain of HMGA2 was detected in 68 out of 180 samples and is known to upregulate IGF2BP2. Additionally, studies have shown that larger fibroids tend to express higher levels of the IGF-1 growth factor.18
IGFBP2 targets HMGA2, a gene involved in the etiology of mesenchymal malignancies and the 2nd most frequently expressed gene in HMGA2 subtypes. While IGFBP2, HMGA2 and IGF2 are generally not expressed in adult tissues, ADAM12 shows specific upregulation, and PAPPA2 is identified as the most abundant subtype associated with HMGA2. Additionally, IGF2 can bind to the IGF-1 receptor, promoting cellular growth.5
MED 12 mutations
The MED12 and COL4A5/6 genes also play significant roles in the development of UL. MED12 mutations represent one of the most common genetic abnormalities in UL, accounting for approx. 70% of UL cases and 80–90% of all UL. These mutations are also associated with increased expression of the RAD51B gene. Furthermore, MED12 mutations have been identified in 10–20% of leiomyosarcomas. Functionally, MED12 mutations disrupt transcriptional repression, impacting the WNT signaling pathway and key cell cycle regulators. This results in reduced expression of REST and increased activity of mTORC, contributing to tumor growth.27
Chromosome 14
The development of UL has also been associated with 3 key genes: ESR2 (estrogen receptor β), TGF-β and RAD51B. Although ESR2 is located in proximity to these genes, its precise role in UL pathogenesis and the frequency of related abnormalities remain unclear. TGFB3 has been shown to promote the expression of ECM components, including COL1A1 and FN1, while TGFB1 is associated with increased fibromodulin expression.10 HMGA2 frequently selects RAD51B as a translocation partner, although HMGA1 and HMGA2 rearrangements generally involve distinct genomic regions. RAD51B, which encodes a double-strand DNA break repair enzyme, may contribute to genomic instability when disrupted.5
Chromosome 17
One gene located on chromosome 17 – TP53 (TSG) and 1 enzyme 17β-hydroxysteroid dehydrogenase – have been implicated in the pathogenesis of UL. TP53 exhibits chromosomal loss in 2 out of 6 analyzed samples, while 17β-hydroxysteroid dehydrogenase is frequently hypermethylated in UL tissues. Such hypermethylation may suppress TSG activity, thereby contributing to tumor development. Furthermore, the involvement of estrogen signaling and the KRT19 pathway has also been suggested.24
Chromosome 22
Our research identified 4 genes located in close proximity on chromosome 22, including PDGFB and TNRC6B. Overexpression of PDGFB has been associated with the development of ULs, and mutations in this gene have also been linked to ovarian cancer. DEPDC5 has been reported to harbor a truncating mutation, while SMARCB1 exhibits a rearrangement that results in the biallelic loss of its tumor suppressor function.25
Chromosome X
The Xp11 and Xq26 regions of the X chromosome are particularly susceptible to hypomethylation-related epigenetic alterations, which may contribute to tumorigenesis.8
Discussion
A multifaceted approach has been employed to investigate the causes of UL, focusing on the interplay between genetics, hormones and growth factors (GFs) in their pathophysiology.25 Multiple attempts have been made to identify the abnormal genetic etiology of UL. Genetic abnormalities and aberrations are the primary focus of fibroid research as an explanation for their emergence. Chromosomal gains, losses, rearrangements, SNPs, and translocations are the most common genetic aberrations associated with the development of UL. Among these, deletions on chromosome X and 7q, as well as rearrangements of 12q15, 6p21 and 10q22, are the most frequent.28
The relevance of genetics in UL is based on several key principles: risk assessment, early disease detection, personalized treatment approaches, fertility counseling, prognosis, and research. High-risk patients, particularly those with a personal or family history of UL and those of African descent, may benefit from early detection and management. This proactive approach can help minimize the risks associated with delayed or complicated surgeries and support fertility-sparing treatments. Additionally, genetic information can guide treatment decisions, including the selection of personalized therapies and influencing responses to certain medications. Prognostic information regarding the behavior and progression of UL can help guide clinicians and patients in making informed decisions about management and treatment options. Lastly, understanding the genetic basis of UL can drive further research into their underlying causes, molecular mechanisms and the development of potential targeted therapies.
The significance of epigenetics in the formation of ULs is still being investigated, and the literature does not yet fully characterize this involvement. The interaction between genetics and epigenetics may help explain why certain genes and phenotypes are more prevalent in UL. Numerous alterations appear to occur at chromosomal loci strongly associated with their development. Other loci, including proto-oncogenes, growth factors and TSGs, have also been shown to undergo several epigenetic modifications.12 For each type of modification, epigenetics must be further studied in terms of statistical significance and population samples. Lastly, further research is needed to determine how changes in the size and architecture of UL are related to these investigations.
Limitations
Not all of the analyzed studies reported sample sizes used to establish genetic associations, leading to inconsistencies across the literature. This heterogeneity limited the ability to directly compare the frequency of genetic variants between studies. As a result, it was not possible to categorize the studies based on the strength or quality of the evidence they provided.
Conclusions
The predominant focus in UL research has been on genetic abnormalities and chromosomal aberrations as key factors in the disease’s etiology. It is estimated that approx. 40% of UL cases are associated with chromosome-specific alterations. The most commonly reported aberrations include chromosomal gains, losses, rearrangements, SNPs, and translocations. Among these, deletions on chromosome X and 7q, as well as rearrangements involving 12q15, 6p21 and 10q22, are the most recurrent. One gene that has emerged as particularly significant in the pathogenesis of UL is MED12, which has been frequently implicated in tumor development. However, inconsistencies across studies limit the ability to directly compare genetic associations, warranting cautious interpretation of the findings.
Supplementary data
The supplementary materials are available at https://doi.org/10.5281/zenodo.17223641. The package includes the following files:
Supplementary Fig. 1. Genes most strongly associated with the pathogenesis of ULs.
Use of AI and AI-assisted technologies
Not applicable.