
















Abstract
Background. Alpha-solanine (α-solanine) is the main glycoalkaloid in potato plants. It possesses anticarcinogenic properties and exerts toxic effects. Alpha-solanine can regulate the nuclear factor kappa B (NF-κB) signaling pathway in cancer cells and macrophages. However, little is known about the anti-inflammatory effects and the related molecular mechanisms of α-solanine on endothelial cells.
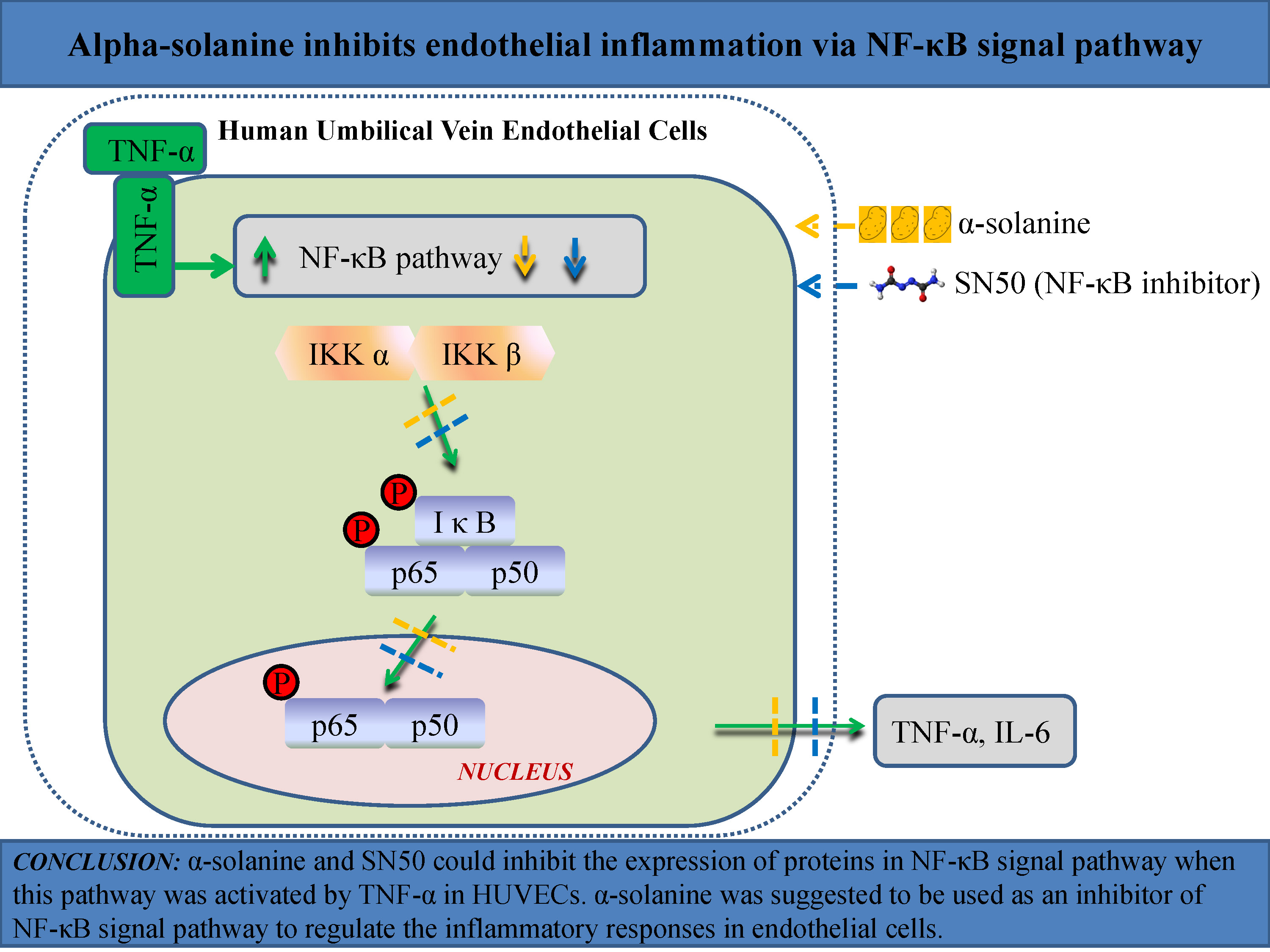
Objectives. This study aims to examine the effects of α-solanine on endothelial inflammation in vitro, and to evaluate its influence on regulating the NF-κB signaling pathway.
Materials and methods. Tumor necrosis factor alpha (TNF-α)-pcDNA3.1(+) plasmid vector was constructed and transfected into human umbilical vein endothelial cells (HUVECs). The expression of TNF-α was examined with quantitative reverse transcription polymerase chain reaction (qRT-PCR) and western blot. Following treatment with α-solanine or the specific NF-κB inhibitor SN50 for 24 h, cell viability was detected using Cell Counting Kit-8 (CCK-8) assay. Interleukin 6 (IL-6) and TNF-α levels in cell supernatant were detected using enzyme-linked immunosorbent assay (ELISA). The relative protein levels of phospho-P65 (p-P65), phospho-inhibitor of NF-κBα (p-IκBα) and IκB kinase (IKK) α/β were examined with western blot.
Results. The α-solanine inhibits TNF-α-induced inflammatory injury in HUVECs. Compared with control cells, the cell viability was significantly decreased, the levels of TNF-α and IL-6 were significantly increased, and the relative protein levels of p-P65, p-IκBα and IKKα/β were significantly upregulated in TNF-α-overexpressed cells. The treatment with α-solanine or SN50 decreased the levels of TNF-α and IL-6, and downregulated the relative protein levels of p-P65, p-IκBα and IKKα/β in TNF-α-overexpressed HUVECs.
Conclusions. This study demonstrated for the first time that α-solanine inhibits endothelial inflammation through the NF-κB signaling pathway. The α-solanine was suggested to be an inhibitor of the NF-κB signaling pathway in endothelial cells. The anti-inflammatory effect of α-solanine may provide a new perspective for the prevention and treatment of phlebitis.
Key words: TNF-α, NF-κB signaling pathway, inflammatory reaction, α-solanine
Background
Peripheral intravenous infusion is widely used in modern medical practice. Phlebitis is a common complication associated with intravenous therapies.1 The incidence of phlebitis with peripheral intravenous catheter use is 10.5% in hospitalized adult patients in China,1 whereas it is as high as 53% in hospitalized pediatric patients in Jordan.2 Phlebitis is a chemical or mechanical inflammation caused by a long-term intravenous infusion of high-concentration, highly irritant drugs or placement of irritant interventional catheters.1, 3, 4, 5 The routine treatments for phlebitis, such as the external application of magnesium sulfate or mucopolysaccharide polysulfate cream, are unsatisfactory.6, 7, 8 Therefore, developing new strategies for the prevention and treatment of phlebitis caused by intravenous infusion is needed. Potato glycoalkaloids, the naturally occurring steroidal glycoalkaloids, are well known for their toxicity and antimicrobial activity.9, 10, 11, 12, 13 Alpha-solanine (α-solanine) is one of the major glycoalkaloids in potato plants.10 Previous studies about α-solanine mostly center on its anticarcinogenic properties and toxicant effects.14, 15, 16 It has been reported that α-solanine could promote tumor cell apoptosis, inhibit cell proliferation and suppress angiogenesis.17, 18, 19, 20, 21, 22 In addition, α-solanine plays important roles in chemical protection and chemotherapy.23, 24, 25 Shin et al. investigated the anti-inflammatory effect of α-solanine on cultured macrophages in a mouse model of endotoxenia, and the results suggested that α-solanine may be a promising therapeutic agent against inflammatory diseases.26
It has been indicated that the role of α-solanine in cancer cells and macrophages is associated with the nuclear factor kappa B (NF-κB) signaling pathway.26, 27, 28 It is a prototypical pro-inflammatory signaling pathway that can be activated by various cytokines, including tumor necrosis factor alpha (TNF-α).29 The NF-κB family consists of 5 proteins: p65, cRel, RelB, p50, and p52. Among them, p65 possesses a transactivation domain and therefore can affect transcription. In resting cells, NF-κB proteins exist in the cytoplasm as inactive forms by binding to their inhibitory proteins IκBs. The IκBα is the prototypical member of the IκB family. The activation of NF-κB depends on IκBα phosphorylation. The IκBα phosphorylation can stimulate ubiquitination and proteasomal degradation, leading to the nuclear translocation of liberated NF-κB, and subsequently the transcription of inflammatory molecules. The phosphorylation of IκB proteins is regulated by IκB kinases (IKK): IKKα and IKKβ.30, 31 Dysregulated NF-κB signaling can cause an imbalance of immune homeostasis, leading to autoimmune diseases, chronic inflammatory diseases and cancers.32, 33, 34 In addition, the activation of the NF-κB signaling pathway is involved in the inflammation-induced activation of the tryptophan-kynurenine metabolic system and mitochondrial impairment, which is implicated in neurodegenerative diseases and psychiatric disorders.35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45 The SN50 is a specific NF-κB inhibitor that could suppress the translocation of the NF-κB active complex into the nucleus.46, 47 It has been reported that SN50 can attenuate inflammation in lung injury and traumatic brain injury.48, 49
The anti-inflammatory effect of α-solanine has been reported in macrophages; however, the effect of α-solanine on endothelial inflammation and the related mechanisms have not been reported until now. Elucidating the effects of α-solanine on endothelial inflammation in vitro as well as evaluating its influence on the NF-κB signaling pathway could provide an experimental basis for the clinical management of phlebitis.
Objectives
This study aims to examine the effects of α-solanine on endothelial inflammation in TNF-α-overexpressed human umbilical vein endothelial cells (HUVECs). Furthermore, whether α-solanine regulates the TNF-α-induced NF-κB signaling pathway was explored.
Materials and methods
Cells and transfection
The HUVECs (BNCC249736) were provided by Beijing Beina Chuanglian Institute of Biotechnology (Beijing, China) and cultured in Endothelial Cell Medium (1001; ScienCell™, Carlsbad, USA). The cells were incubated at 37°C in a humidified atmosphere of 5% CO2. The α-solanine (20562-02-1) was purchased from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China), and SN50 (HY-P0151) from MedChemExpress (Monmouth Junction, USA). The pcDNA3.1(+) (P0157) was provided by Wuhan Miaoling Biotechnology Co., Ltd. (Wuhan, China). Before transfection, the TNF-α overexpression vector (TNF-α-pcDNA3.1(+) plasmid vector) was prepared. The pcDNA3.1(+) plasmid vector was digested with XhoI (ER0691; Thermo Fisher Scientific, Waltham, USA). The sequences of the TNF-α gene were amplified and inserted into the pcDNA3.1(+) vector. The ligated vector was transformed into Trans5α Chemically Competent Cell (CD201-01; TransGen Biotech Co., Ltd.) and the positive clones were selected. The TNF-α-pcDNA3.1(+) and pcDNA3.1(+) plasmids were extracted using the Endo-free Plasmid Mini Kit II (D6950-01; Omega Bio-Tek, Norcross, USA) and verified through enzyme digestion. Cell transfection was performed using Lipofectamine 3000 (L3000015; Thermo Fisher Scientific).50, 51 The HUVECs were seeded in 6-well plates at a density of 5×105 cells per well and allowed to grow to 70% confluence. Then, the cells were incubated in fresh medium without serum. Next, 5 μL Lipofectamine 3000 was diluted with 125 μL opti-MEM medium and incubated with the diluted plasmid (2.5 μg plasmid DNA diluted in 125 μL opti-MEM medium (Thermo Fisher Scientific) and 5 μL Lipofectamine 3000) for 15 min at room temperature. The HUVECs were incubated with 250 μL DNA-liposome complexes (containing 10 µL Lipofectamine 3000 and 2.5 µg plasmid DNA) at 37°C for 4 h, and then the medium was refreshed. At 48 h following transfection, the cells were treated with 10 μg/mL α-solanine or 5 mg/mL SN50 for 24 h.52, 53 Cells transfected with the empty plasmid vector pcDNA3.1(+) were called the vector group.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Ultrapure RNA Kit (CW0581M; CWBIO, Beijing, China) was used for RNA extraction. The RNA concentrations were measured under an ultraviolet spectrophotometer (NP80, NanoPhotometer; Implen GmbH, Munich, Germany). Reverse transcription was performed to synthesize cDNA using a commercial kit (R223-0; Vazyme Biotech Co., Ltd., Nanjing, China) and qRT-PCR was performed with SYBR Green reagents (A4004M; LifeInt, Xiamen, China). The reaction system contained 1 μL cDNA, 10 μL 2×SYBR Green PCR Master Mix, 0.4 μL upstream and downstream primers, and 8.2 μL dH2O without RNase. Cycling parameters were 10 min at 95°C for initial denaturation, then 40 cycles of 10 s at 95°C for denaturation, 30 s at 58°C for annealing, and 30 s at 72°C for extension. The primer sequences of TNF-α were 5’-CGAGTGACAAGCCTGTAGCC-3’ (upstream) and 5’-TGAAGAGGACCTGGGAGTAG-3’ (downstream). The primer sequences of β-actin were 5’-TGGCACCCAGCACAATGAA-3’ (upstream) and 5’-CTAAGTCAGATTCCGCTAGAGAAGCA-3’ (downstream). Relative TNF-α mRNA expression levels were calculated using the 2−ΔΔCq method.54 The β-actin was used as the housekeeping gene.
Western blot
The cells were lysed with radioimmunoprecipitation assay (RIPA) Lysis Buffer (C1053; Applygen Technologies Inc., Beijing, China) and incubated on ice for 20 min. After centrifugation at 12,000 rpm for 10 min, the concentration of proteins in the supernatant was determined using a bicinchoninic acid (BCA) kit. Equal amounts of protein were separated using sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by transferring onto a polyvinylidene fluoride (PVDF) membrane (IPVH00010; Millipore, Boston, USA). The membrane was blocked in nonfat milk and incubated overnight with the specific antibody at 4°C. Mouse monoclonal anti-actin (TA-09) was purchased from Zhongshan Jinqiao Biotechnology Co., Ltd. (Beijing, China). Rabbit anti-P65 (10745-1-ap) and rabbit anti-inhibitor of NF-κB (anti-IκBα) (10268-1-ap) were obtained from Proteintech Group Inc. (Chicago, USA). Rabbit anti-phospho-P65 (af2006), rabbit anti-IκB kinase (IKK) α/β (af6014), and rabbit anti-phospho-IκBα (af2002) were purchased from Affinity Biosciences (Cincinnati, USA). These antibodies were used at a dilution of 1:1000. After washing with Tris-buffered saline with Tween 20 (TBST), the membrane was incubated with the secondary antibody, including peroxidase-conjugated goat anti-mouse IgG (H+L) (ZB-2305) and peroxidase-conjugated goat anti-rabbit IgG (H+L) (ZB-2301) at a 1:2000 dilution for 2 h at room temperature. These secondary antibodies were from Zhongshan Jinqiao Biotechnology Co., Ltd (Beijing, China). The signals were visualized by the hypersensitive luminescent solution (RJ239676; Thermo Fisher Scientific) with a chemiluminescence imaging system (ChemiDoc™ XRS+; Bio-Rad, Hercules, USA).
CCK-8 assay
Cell Counting Kit-8 (CCK-8) assay was performed to examine cell viability. This assay uses a tetrazolium salt WST-8 (water-soluble tetrazolium 8), which produces the orange-colored formazan when 2 electrons are received from viable cells through an electron mediator, 1-Methoxy phenazinium methylsulfate (PMS), by nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADPH). The amount of formazan is dependent on the number of living cells. After washing with phosphate-buffered saline (PBS), 5×103 cells were plated into the 96-well plate. The α-solanine was dissolved in absolute ethanol. The next day, 1, 3, 6, 9, or 12 μL of α-solanine (1 mg/mL) were added to treat the cells for 24 h. The medium was then refreshed and 10 μL of CCK-8 was added into the wells and incubated for 2 h at 37°C. Optical densities were measured with an automatic microplate reader (WD-2102B; Beijing Liuyi Biotechnology Co., Ltd., Beijing, China) at a wavelength of 450 nm.
ELISA
The TNF-α and IL-6 concentrations in cell supernatant were measured with the Human TNF-α enzyme-linked immunosorbent assay (ELISA) Kit (MM-0122H1; Jiangsu Meimian Industrial Co., Ltd., Yancheng, China) and Human IL-6 ELISA Kit (MM-0049H1; Jiangsu Meimian Industrial Co., Ltd.), respectively, following the manufacturer’s protocols. Briefly, 50 μL standards with different concentrations or 50 μL samples were added to the wells. Then, 100 μL of enzyme-labeled reagents were added to each well, except for the blank well, and incubated at 37°C for 60 min. After washing with washing buffer 5 times, 50 μL substrates A and B were added and the plates were incubated in the dark at 37°C for 15 min. Then, 50 μL termination solution was added to each well and the optical density (OD) values at a wavelength of 450 nm were measured within 15 min.
Statistical analyses
The IBM SPSS v. 20.0 software (IBM Corp., Armonk, USA) was used to analyze statistical data. Each sample was selected independently. Kruskal–Wallis H test (K–W test) followed by Dunn–Bonferroni test (D–B test) was performed to compare the differences between multiple groups. All the statistical values of pairwise comparisons between the groups using D–B test for post hoc testing are shown in Table 1. Statistical significance was defined as p < 0.05.
Results
Effect of different concentrations of α-solanine on the viability of HUVECs
Different concentrations of α-solanine were used to treat HUVECs for 24 h. Then, the cell viabilities were measured using the CCK-8 assay. As shown in Figure 1A, HUVEC viability was significantly decreased by treatment with 30 μg/mL, 60 μg/mL, 90 μg/mL, and 120 μg/mL α-solanine (K–W test: H = 44.802, df = 5, p < 0.001). Cell viability was not affected by treatment with 10 μg/mL α-solanine (D–B test, p = 1.000). Therefore, we chose α-solanine at the concentration of 10 μg/mL to conduct the following experiments.
Effect of α-solanine on cell viability
of TNF-α-overexpressed HUVECs
The TNF-α overexpression vector was constructed and transfected into HUVECs, and TNF-α expression levels in HUVECs were examined with western blot and qRT-PCR. The results in Figure 1B show that, compared with the TNF-α negative control vector (pcDNA3.1(+) plasmid vector)-transfected cells, both the TNF-α relative mRNA expression level (D–B test, p = 0.048) and the TNF-α relative protein expression level (D–B test, p = 0.010) in the TNF-α overexpression vector-transfected cells were significantly increased. The CCK-8 assay demonstrated that, compared with the vector group, cell viability was significantly decreased in the TNF-α group (D–B test, p = 0.009). The difference in cell viability between the TNF-α + α-solanine group and the TNF-α group was not significant (D–B test, p = 1.000) (Figure 1C).
Effect of α-solanine on TNF-α
and IL-6 protein levels
in TNF-α-overexpressed HUVECs
An ELISA was used to detect TNF-α and interleukin 6 (IL-6) protein levels in the cell supernatant of HUVECs. The results presented in Figure 2 show that, compared with the vector group, TNF-α (D–B test, p < 0.001) and IL-6 protein levels (D–B test, p < 0.001) were significantly increased in the TNF-α group. In TNF-α-overexpressed cells, α-solanine significantly decreased the elevated protein levels of TNF-α (D–B test, p = 0.015) and IL-6 (D–B test, p = 0.001).
Effect of α-solanine on phospho-inhibitor of NF-κBα (p-IκBα), phospho-P65 (p-P65) and IKKα/β relative protein expression levels in TNF-α-overexpressed HUVECs
The p-IκBα, p-P65 and IKKα/β relative protein expression levels in TNF-α-overexpressed HUVECs were determined using western blot assay. We found that the relative protein expression levels of p-IκBα (D–B test, p < 0.001), p-P65 (D–B test, p = 0.001) and IKKα/β (D–B test, p = 0.018) were significantly increased in the TNF-α group compared with the vector group. However, in TNF-α-overexpressed HUVECs, the increased p-IκBα (D–B test, p = 0.032), p-P65 (D–B test, p = 0.021) and IKKα/β (D–B test, p = 0.001) relative protein expression levels were significantly suppressed by treatment with α-solanine (Figure 3).
Effect of SN50 on cell viability,
as well as IL-6 and TNF-α levels
in TNF-α-overexpressed HUVECs
The SN50 is a specific NF-κB inhibitor that could suppress the translocation of the NF-κB active complex into the nucleus. To evaluate whether α-solanine has a similar function to that of SN50, the cells were treated with 5 mg/mL SN50 for 24 h. As shown in Figure 4A, SN50 did not affect the cell viability of TNF-α-overexpressed HUVECs (D–B test, p = 1.000). As shown in Figure 4B, both the TNF-α level (D–B test, p < 0.001) and the IL-6 level (D–B test, p = 0.005) were significantly increased in TNF-α-overexpressed HUVECs; however, SN50 treatment suppressed the elevated TNF-α (D–B test, p = 0.002) and IL-6 (D–B test, p < 0.001) levels.
Effect of SN50 on p-IκBα, p-P65
and IKKα/β relative protein expression levels in TNF-α-overexpressed HUVECs
The results presented in Figure 5 demonstrate that p-IκBα (D–B test, p = 0.001), p-P65 (D–B test, p = 0.043) and IKKα/β (D–B test, p = 0.001) relative protein expression levels were increased in the cells transfected with the TNF-α vector compared with the cells transfected with the pcDNA3.1(+) vector. The SN50 treatment significantly suppressed the increased p-IκBα (D–B test, p = 0.048), p-P65 (D–B test, p = 0.041) and IKKα/β (D–B test, p = 0.004) relative protein expression levels in TNF-α-overexpressed HUVECs.
Discussion
The present study demonstrated the effects of α-solanine on inflammation in HUVECs. The α-solanine inhibited the expression of TNF-α and IL-6 in TNF-α-overexpressed HUVECs, and these effects were related to the suppression of the NF-κB signaling pathway.
Phlebitis is an inflammation of the vein. Vascular endothelial cells make up the innermost surface of blood vessels. Inflammatory injury and apoptosis of endothelial cells are observed in many human diseases, including phlebitis.55, 56, 57 In the present study, TNF-α was overexpressed in HUVECs to establish a model of venous endothelial inflammation. Consistent with the previous report revealing that TNF-α can induce inflammatory injury,58 our study showed that TNF-α increased IL-6 expression, activated the NF-κB signaling pathway and inhibited the proliferation of HUVECs. These results demonstrated that TNF-α can induce inflammation injury in endothelial cells.
A large number of studies have confirmed the toxic effects of α-solanine on cancers.14, 15, 16, 23, 24 Only a limited number of studies investigated the anti-inflammatory effect of α-solanine on macrophages and in endotoxemia model mice.26, 59 Kenny et al. reported that α-solanine possesses anti-inflammatory properties in lipopolysaccharide (LPS)-induced Raw264.7 macrophages59 by reducing the production of nitric oxide (NO) and decreasing the levels of pro-inflammatory cytokines. This study provided more evidence for the anti-inflammatory effect of α-solanine. We investigated the anti-inflammatory effect of α-solanine in endothelial cells. Similar to the effects in LPS-induced macrophages, α-solanine inhibited the expression of the pro-inflammatory mediators TNF-α and IL-6 in TNF-α-overexpressed endothelial cells. The suppression of TNF-α and IL-6 can prevent inflammatory injury and apoptosis of HUVECs.60, 61 Taken together, this evidence suggests that α-solanine can inhibit inflammatory injury of HUVECs. It is noteworthy that the levels of IL-6 and TNF-α in the TNF-α + α-solanine group were significantly higher than in the control group, suggesting that α-solanine may not eliminate the effect of TNF-α overexpression on IL-6 and TNF-α elevation in HUVECs. However, α-solanine could alleviate endothelial inflammation, thus showing a positive role in the treatment of phlebitis.
The NF-κB pathway is considered the most typical inflammatory pathway, and many inflammatory mediators are found downstream of NF-κB.62, 63 The study by Lu et al. showed that α-solanine downregulates the nuclear content of NF-κB in human melanoma cells.28 In human hepatocellular carcinoma HepG2 cells, α-solanine-induced oxidative stress is associated with the NF-κB pathway.27 Natural or synthetic NF-κB inhibitors could prevent LPS-induced injury in endothelial cells by decreasing the levels of inflammatory factors.64, 65 In the current study, we investigated the effect of α-solanine on the NF-κB signaling pathway in endothelial cells and found that α-solanine inhibited TNF-α-induced NF-κB signaling pathway activation in HUVECs by decreasing p-IκBα, p-P65 and IKKα/β relative protein expression levels. Our results were consistent with the reports on LPS-induced macrophages and endotoxemia model mice which demonstrated that α-solanine abrogates inflammatory responses by inhibiting the NF-κB signaling pathway. We found that p-IκBα was not significantly decreased in the TNF-α + α-solanine group compared with the control group. However, p-IκBα was significantly decreased in the TNF-α + α-solanine group compared with the TNF-α group. These results indicate that α-solanine may not eliminate the effect of TNF-α overexpression on IκBα phosphorylation in HUVECs. However, α-solanine shows an inhibitory effect on the phosphorylation of IκBα in TNF-α-overexpressed HUVECs. Taken together, this evidence suggests that α-solanine protects against inflammatory diseases via NF-κB suppression. Previous studies have shown other molecules and signaling pathways that were regulated by α-solanine, such as matrix metalloproteinases (MMPs), Akt/mTOR signaling pathway, STAT3 signaling pathway, and ERK and PI3K/Akt signaling pathways.20, 28, 66, 67, 68 Whether these molecules and signaling pathways are involved in the effect of α-solanine on protecting against endothelial inflammation is still unknown. The molecular mechanisms underlying the effect of α-solanine on endothelial inflammation need to be further explored.
In the present study, we found that the effects of α-solanine on TNF-α-overexpressed HUVECs seem to be similar to that of SN50. The activation of the NF-κB pathway leads to the nuclear translocation of NF-κB protein and the activation of downstream genes.69 The SN50 is a specific NF-κB inhibitor that could suppress the translocation of the NF-κB active complex into the nucleus. We demonstrated that both α-solanine and SN50 could decrease the relative expression levels of proteins in the NF-κB signaling pathway, such as p-IκBα, p-P65 and IKKα/β. These results suggested that α-solanine can act as an NF-κB inhibitor in the process of inflammatory injury in HUVECs. However, we did not examine the nuclear translocation of NF-κB proteins. To confirm the possibility of α-solanine as an NF-κB inhibitor, a study investigating the effect of α-solanine on NF-κB nuclear translocation will be carried out in the future.
This study demonstrated the inhibitory effect of α-solanine on endothelial inflammation in vitro. Further studies need to be performed on phlebitis animal models and phlebitis patients to confirm the anti-inflammatory effect of α-solanine in vivo. Expanding the application of α-solanine on other inflammatory diseases may be a good research direction in the future. Potato glycoside alkaloids are a group of easily obtained natural steroid compounds. Studies on the function of α-solanine in inflammation will provide a new perspective for the prevention and treatment of phlebitis, as well as new ideas for the research and development of potato glycoside alkaloids.
Limitations
There are 3 major limitations to this study. First, we only investigated the anti-inflammatory effects of α-solanine in vitro. The therapeutic effect of α-solanine on phlebitis in vivo needs to be confirmed on animal models in our further studies. Second, to fully elucidate the effect of α-solanine on inflammatory response in HUVECs, more inflammatory factors and mechanisms that are related to inflammation need to be examined. Third, TNF-α-transfected cells are not a good model of inflammation. The use of endotoxin or inflammatory agents will better induce inflammation.
Conclusions
This is the first in vitro study demonstrating that α-solanine inhibits endothelial inflammation through the NF-κB signaling pathway. The findings support the potential application of α-solanine for the clinical management of phlebitis and describe the molecular mechanism underlying the protective function of α-solanine against endothelial inflammation.




