Abstract
Anemia is a common finding among patients with liver diseases. Patients who suffer from anemia are at a higher risk of liver function decompensation and hospitalization. It affects significantly their quality of life and contributes to mortality. Anemia is present in 70% of patients with liver cirrhosis and with varying incidence accompanies other liver disorders. As the etiology of anemia in liver diseases is multifactorial, various cases represent different clinical entities. Anemia accompanying hepatic disorders can be broadly divided into several types, such as anemia associated with blood loss, as well as aplastic, hemolytic and micronutrient deficiency anemia. However, it is sometimes difficult to delineate between those types in the clinical practice, as several pathophysiological causes can be present in one patient. It is reported that the most common cause of anemia in liver disease is blood loss and iron deficiency. Still, the incidence of unclear cases reaching over 50% suggests that other types of anemia can be underdiagnosed. This review comprehensively describes less frequent types of anemia associated with liver disease, namely hemolytic and aplastic anemia (AA). Hemolytic anemia can complicate autoimmune liver diseases or be a manifestation of membranopathy of red blood cells, dependent on severe hepatic function impairment or alcoholic liver disease. Aplastic anemia is best known as a sequela of viral hepatitis, but some degree of bone marrow inhibition can complicate virtually all advanced liver diseases.
Key words: liver, anemia, hemolytic, aplastic, hepatology
Introduction
Anemia complicating liver diseases is a common finding in clinical practice that constitutes a significant problem, since it can have unfavorable effects on patient prognosis and quality of life. It is hypothesized that the deleterious effect of anemia is imposed through hypoxia and the promotion of hyperdynamic circulation. Cirrhotic patients with anemia were found to have a higher hospital mortality rate. They also more frequently develop complications of cirrhosis such as type 2 hepatorenal syndrome, and supposedly gastrointestinal bleeding and ascites.1, 2
Anemia prevalence in hepatology is associated with the degree of impairment of liver function and portal hypertension. Prevalence of anemia is especially high in the context of liver cirrhosis, where decreased concentration of hemoglobin is reported to affect around 70% of patients.3, 4 As reported by Scheiner et al., more severe cases of anemia are less common. In their retrospective analysis, they found out that moderate to severe anemia was present in 28% of chronic liver disease cases.4 Noticeably, in most cases, the etiology of anemia remains unclear. Anemia of unknown origin constituted 53% of all cases in Scheiner’s cohort, followed by bleeding (25%) and iron deficiency (9%).4 Unfortunately, the high prevalence of anemia can lead to a misconception that it is an essential feature of liver disease. In effect, less obvious reasons for anemia among patients suffering from hepatic disease tend to be overlooked or diagnosed with a significant delay. Both hemolytic and aplastic anemia (AA) are rare complications of liver disease, but clinicians should be aware of them due to the serious prognosis.
According to World Health Organization’s (WHO) guideline, the thresholds for the diagnosis of anemia are set at 13 g/dL for men and 12 g/dL for women. For moderate and severe anemia, the cutoffs are <11 g/dL and <8 g/dL, respectively.5
Hemolytic anemia manifests itself as shorter than the normal lifespan of erythrocyte. A number of different classifications of hemolytic anemia have been implemented. The broadest, but also, due to its clinical implications, the most widely used, is a division depending on the involvement of immune-mediated mechanisms of hemolysis. The clinical picture being the most suggestive of hemolysis, can be described as an increased bilirubin concentration, high lactate dehydrogenase blood activity and a low concentration of haptoglobin in the presence of normocytic anemia with reticulocytosis.6
Aplastic anemia is a rare clinical entity defined as an injury to precursor hematopoietic cells. The pathomechanism of AA is considered immune-mediated, but antigens triggering the response are not fully characterized. Cytopenia tends to occur in all 3 lines of blood cells. To diagnose AA, the thresholds were established for hemoglobin at 10 g/dL, platelets at 50,000/μL and neutrophils at 1500/μL. For the diagnosis, however, bone marrow examination is necessary, as it can show hypocellularity in the absence of bone marrow infiltration or fibrosis.7
Objectives
The aim of this paper is to summarize the available information about the rare causes of anemia – mainly hemolytic and aplastic – among patients with liver disease. As research on that topic is scattered, a review article seems to be the best choice to give a theoretical basis for medical practitioners who face difficulties diagnosing anemic patients with liver disorders.
Materials and methods
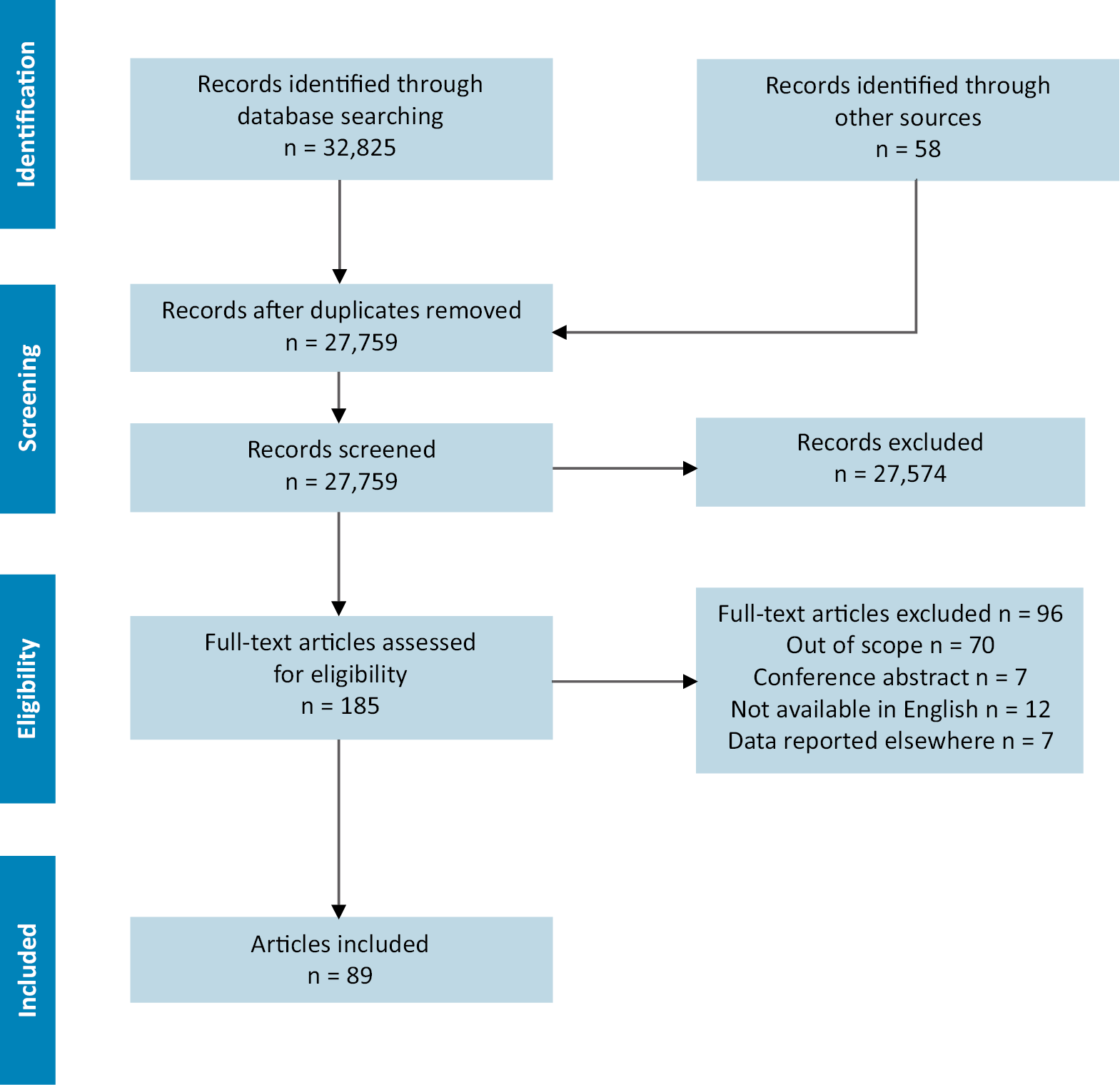
A selection of available literature in PubMed, Cumulative Index to Nursing and Allied Health Literature (CINAHL) and Cochrane Library databases was performed in July 2021. The search included both original and review articles. The utilization of search terms, such as “liver disease”, “liver cirrhosis”, “hepatitis”, “cholestasis”, “liver injury”, “alcoholic liver disease”, “aplastic”, “hypoplastic”, “hemolytic”, “autoimmune”, “anemia”, “hemolysis”, “acanthocytosis”, and “bone marrow aplasia” let us identify 32,825 articles. Titles and abstracts were screened from that number, which limited the number of applicable papers to 3,726 original articles and 127 review articles. We included studies that focused on hemolytic and aplastic anemia in patients with liver diseases. Studies published in other languages than English, without the abstracts available online, as well as duplications were excluded. Citations and reference lists of selected articles were analyzed in the next step, allowing to identify 185 articles for a full-text review (Figure 1). Articles screening was performed independently by both authors, and in case of conflicting opinions papers were discussed on a case-by-case basis.
Hemolytic anemia
Compared with bleeding or micronutrient deficiency, hemolysis is an uncommon cause of anemia in patients with liver disease. The possible mechanisms of hemolysis include immune-mediated destruction of red blood cells with the involvement of antibodies, or non-immune mechanisms, dependent mainly on acquired structural aberrations of red blood cells.
Antibody-mediated hemolysis is one of the main mechanisms of the acquired cases of hemolytic anemia in the general population. A well-described feature of liver and bile duct autoimmune diseases has a high rate of coexistence with other autoimmune diseases. In the case of autoimmune hepatitis, as many as 20–50% of patients have other autoimmune disease. Those numbers can be even higher when primary biliary cholangitis (PBC) is considered, rising up to 84%, as reported by Culp et al.8, 9 Autoimmune hepatitis, PBC and primary sclerosing cholangitis (PSC) significantly differ from each other in the incidence of various extrahepatic autoimmune diseases. However, autoimmune hemolytic anemia (AIHA), characterized by a positive direct Coombs test, remains a rare comorbidity for all three. The reported prevalence of AIHA among AIH patients is less than 1%.9 The diagnosis of AIHA complicating PBC or PSC course is even less frequent.10, 11 In fact, it is not currently clear if AIHA associated with PBC and PSC represents a distinct clinical entity or is just a coexistence of 2 independent diseases by chance. In the context of AIHA complicating autoimmune liver disease, standard treatment regimens are usually recommended. The clinical guidelines on the management of secondary AIHA, available from 2017, do not specifically discuss autoimmune liver conditions.12 The first-line treatment includes corticosteroids and immunosuppressants, like azathioprine, cyclophosphamide or cyclosporine.10 In cases with no satisfying improvement on corticosteroids, rituximab is a recommended option, and in the context of a limited access to rituximab, splenectomy should be considered.13 It should, however, be noted, that the effectiveness of monotherapy with ursodeoxycholic acid (UDCA) has been described in mild cases of AIHA associated with PBC.14
A major portion of hemolytic anemia cases complicating liver disease is not dependent on immune-mediated mechanisms. Severe acute or chronic liver injury can lead to red blood cell membrane alternations and, in consequence, to a shortened life span of erythrocytes. The liver is an organ playing a crucial role in lipid metabolism – the insufficiency of liver functions causes lipid disturbances in cell membranes. An increasing amount of cholesterol in the erythrocyte cell membrane results in the enlargement of its surface – an effect observed as macrocytosis on complete blood count.15 Moreover, echinocytosis and stomatocytosis of red blood cells can be an effect of phosphatidylcholine alternation, which in turn is a result of liver disease.15 In more severe cases of liver disease, spur cell anemia (acanthocytosis) can develop. Spur cell anemia is a rare complication of liver disease, but its association with the liver function impairment is well described in the literature. Spur cell anemia especially often accompanies cases of alcoholic liver disease.16 The name of the disease is derived from the characteristic morphology of erythrocytes, which are enlarged and develop thorny-like processes. The primary mechanism behind spur cell anemia are changes in cholesterol to phospholipid ratio. In effect, the erythrocyte cell membrane loses its normal elastic properties. Deformed erythrocytes become prone to sequestration and destruction by macrophages in spleen. Interestingly, the aberration in lipid cell membrane composition is clearly acquired; red blood cells that were transfused to cirrhotic patients tend to gradually change their lipid composition and have a shortened lifespan.17 Data from pediatric cohorts suggest that besides cell membrane lipid disturbances, vitamin E deficiency can play a major role in the pathogenesis of hemolytic anemia in the context of liver disease.18
The risk of spur cell anemia development is 2 times higher in women with alcoholic liver disease.16 Patients who, besides alcoholic liver disease, have other comorbidities, especially chronic obstructive pulmonary disease (COPD), are at a higher risk of spur cell anemia. Tariq et al. hypothesize in their study that COPD and alcoholic liver disease can have common destructive mechanisms of action towards red cell membranes through increased reactive oxygen species (ROS) production in both diseases.16
Spur cell anemia is associated with a poor prognosis and a reported average survival time of 1 year.19 Scarce reports suggest that liver transplantation can play a curative role in both liver disease and spur cell anemia.20 Data on splenectomy usefulness in spur cell anemia treatment are insufficient. Early data showed that spur cells transferred to healthy asplenic recipients had normal survival.17 However, decreasing spleen blood flow by placing a transjugular intrahepatic portosystemic shunt (TIPS) has not been observed to affect the disease course positively.20
Zieve’s syndrome represents another clinical entity, which essential feature is non-immune mediated hemolytic anemia, strongly associated with alcoholic liver disease. The syndrome is rare, and only several hundred cases have been reported so far; however, it seems to be underreported due to a limited awareness of clinicians.21 Zieve’s syndrome, besides transient hemolytic anemia, is characterized by jaundice and hyperlipidemia. Hemolytic anemia associated with Zieve’s syndrome has similar pathomechanism to spur cell anemia in cases of chronic liver failure. The most important distinctive feature of hemolytic anemia associated with Zieve’s syndrome is its temporary character. Significant disturbances that are believed to play a role in the pathogenesis of hemolysis in Zieve’s syndrome are lysolecithin and lysocephalin induction, and their accumulation in red cell membranes – changes which are dependent on vitamin E deficiency.21 The symptoms of Zieve’s syndrome tend to wear off after several weeks (usually 4–6). Since this syndrome is usually caused by excessive alcohol consumption, abstinence can shorten the time to subsiding of symptoms. Due to the rarity of this clinical entity, well-designed studies concerning optimal treatment are lacking. The awareness of Zieve’s syndrome can have important clinical implications, as hemolysis resulting from it can influence the Maddrey score, prompting the initiation of corticosteroid treatment. Because of the fact that Zieve’s syndrome is believed not to be an immune-mediated type of hemolysis, glucocorticoid therapy is regarded of little value, and may result in an increased risk of iatrogenic complications.21
Non-immune mediated hemolysis is a common feature of Wilson’s disease (WD), which relatively often can be a presenting symptom leading to diagnosis. Such a course of WD is frequently reported in a younger population.22, 23 The mechanism behind hemolysis in WD is multifactorial. In case of massive necrosis of hepatocytes, a significant load of copper is released to the bloodstream and causes oxidative stress to cell membranes.24 Other possible mechanisms include the sodium pump function impairment and the alternation of cell membrane composition.23 Hemolysis subsides when the pharmacotherapy of WD is introduced, or when the patient undergoes liver transplantation.
Patients with liver disease can also develop anemia due to their medication. The best-documented example of anemia due to medications used in hepatology is ribavirin-induced hemolytic anemia (RIHA). According to the summary of product characteristics, anemia is a very common adverse reaction to ribavirin (>10% of patients), and hemolytic anemia is common (>1% of patients).25 Unfortunately, in the available clinical research, the mechanism of decrease in hemoglobin concentration was not analyzed. A decline of 3 g/dL was observed in 54% of patients taking standard ribavirin dose, and in around 8%, it was greater than 5 mg/dL.26 Data presented in the summary of product characteristics come from the trials of ribavirin combined with peginterferon, which is also known to cause hemolytic anemia by itself. When ribavirin is co-administered with direct-acting antivirals (DAA) instead of peginterferon, RIHA rate ranges between 5% and 40%.27 However, more severe cases of RIHA constitute less than 10% of all cases.28 Exact pathophysiological mechanism of RIHA is unknown. It is postulated that the active form of ribavirin causes cellular shortage of adenosine triphosphate (ATP) in erythrocytes, the consequence of which is impaired glycolysis and oxidative stress, an effect that is dose-related.26, 29 Hemolysis was also observed in other species exposed to ribavirin.30 Reducing a dose of ribavirin or discontinuation is usually an adequate action. The need for blood transfusions is sporadic (0.1%), assuming correct laboratory results monitoring.28
Aplastic anemia
Aplastic anemia is another rare type of anemia, both in the general population and among patients with liver disease. It has been reported to complicate 2% of cases of chronic liver disease,31 but the majority of clinical cases described in literature come from the context of acute liver disease. Aplastic anemia is defined by pancytopenia, accompanied by decreased bone marrow cellularity in the absence of bone marrow infiltration or marrow fibrosis.7 Diagnostic criteria include at least 2 of the following: hemoglobin concentration less than 10 g/dL, platelet count less than 50,000/μL, or neutrophil count below 1500/μL.32 Immune-mediated pathologies are important factors in the development of many AA cases, but very often the exact mechanism of AA remains unclear.
In cirrhotic patients, bone marrow inhibition may contribute to existing anemia. However, anemia itself may not meet the criteria of an aplastic one. Bihari et al. conducted a complex comparison of bone marrow in cirrhotic and control individuals.33 They have found that the number of CD34+ progenitor cells has decreased in advanced cirrhosis. There was a significant correlation of this effect with model for end-stage liver disease (MELD) and Child–Pugh scores, but not with the etiology of cirrhosis. Besides that, the overall cellularity of bone marrow was decreased in cirrhosis; however, some degree of erythroid hyperplasia was observed. The changes in the number of hematopoietic cells can be linked to altered bone marrow microenvironment, with decreased population of niche cells, nerve fibers and Schwann cells, all of which are important in hematopoiesis. On the other hand, the inhibition of hematopoiesis can be linked to an increased concentration of proinflammatory cytokines and a decreased concentration of hematopoietic cell growth factors.33 The awareness of bone marrow functioning impairment led to attempts of setting it as a therapeutic target. A randomized controlled trial, conducted by Anand et al. showed that cirrhotic individuals benefit from therapy with erythropoietin combined with granulocyte colony-stimulating factor. The response was more pronounced in patients in early cirrhosis stages and without a deep depletion of hemopoietic cells in the bone marrow.34
In liver disease, AA is mainly associated with infectious causes. So far, many infectious factors have been linked to AA. The cases of hepatitis-associated aplastic anemia (HAAA) were described after virtually all hepatotropic viral infections, including hepatitis A virus (HAV), hepatitis B virus (HBV), hepatitis C virus (HCV), hepatitis D virus (HDV), and hepatitis E virus (HEV). Besides that, less frequent causes noted in the literature are Epstein–Barr virus (EBV) and cytomegalovirus (CMV) infections. The HBV and HCV acute infections are most common in cases with established causative factor.35 However, the majority of patients are seronegative for known hepatotropic viruses.36 The HAAA associated with nonviral hepatitis, including drug and vaccine-induced hepatitis, has also been reported.37, 38 Distinct clinical picture can be elicited by parvovirus B19 infection, which is a well-known causative factor for the development of AA. It is worth noting that parvovirus B19 has a predilection to erythroid lineage cells; therefore, bone marrow biopsy can show cellularity within reference range, with depletion of erythropoietic cells.39 In fact, an association of parvovirus B19 with AA is much stronger than with hepatitis, but it can cause both clinical entities. Pure red cell aplasia, instead of AA, is also commonly described in the context of HAV infections.
The HAAA was described to accompany 2–5% of hepatitis cases in Western Europe, and 1–5% of AA cases are associated with HAAA.40 It was found to be relatively more common in areas with a high prevalence of human immunodeficiency virus (HIV) infections, suggesting the influence of comorbidities on the occurrence rate of the disease.36 Some authors report higher HAAA prevalence in children than adults, especially among adolescent boys.36 However, other series of cases do not show any association with age or sex, similarly to what is observed in the general AA patients population.41 The HAAA can complicate both mild and severe cases of hepatitis, and the onset of hepatitis preceding HAAA can be both fulminant or insidious.36, 38
The onset of the disease varies, but it is typically observed after 2–3 months after hepatitis.36, 38 However, an earlier onset is not uncommon in literature, and patients can still have laboratory features of acute hepatitis on presentation with HAAA.42 In fact, according to a study in bone marrow transplant patients by Safadi et al., around 40% of patients who develop HAAA still have the laboratory markers of hepatitis on diagnosis.41 At the opposite extreme, HAAA can also be observed in 10% of patients after more than 1 year from a hepatitis episode.41 However, in such a scenario, the causal relationship with previous hepatitis is more controversial.
The reported symptomatology is similar to symptoms of AA in general population, which include fatigue, pallor, bleeding, easy bruising, and infections. Those symptoms can present in various combinations, depending on the degree of damage to particular cell lines, like anemia, thrombocytopenia and leukopenia, and can differ in severity. The diagnosis has to be confirmed by the aplastic or hypoplastic bone marrow in bone marrow biopsy or aspirate examination.
It is suspected that cytotoxic T lymphocytes play a key role in the pathogenesis of HAAA. Ikawa et al. suggest that CD8+ lymphocytes recognize similar antigen patterns of liver and hematopoietic cells.43 In vitro studies have shown that in some cases, the presence of CD8+ lymphocytes in bone marrow correlates with the impairment of cell colonies formation.44 A similar inhibitory effect on colony forming in bone marrow was linked to the increased interferon gamma (IFNγ) concentration,45 which can be produced by CD8+ lymphocytes infiltrating bone marrow. Interferon gamma plays a multidirectional role in bone marrow functioning. It can stimulate bone marrow stem cells in the short term by promoting their differentiation, but in a longer perspective, it plays an inhibitory role by intensifying apoptosis, decreasing the self-renewal of stem cells and disturbing the interaction of stem cells with the bone marrow microenvironment.46
The outcome of HAAA, when untreated, remains fatal in the majority of cases. The HAAA is considered an indication for hematopoietic stem cell transplantation, especially in younger patients with human leukocyte antigen (HLA)-matched siblings. Patients with HAAA are often erythrocyte-dependent and platelet transfusion-dependent, but restrictive thresholds for transfusion should be observed to avoid sensitization before bone marrow transplantation.36 Results of stem cell transplantation from HLA-matched unrelated donors have inferior results.37 When stem cell transplantation is not an available option, or the patient is over 40 years old, the immunosuppressive treatment should be implemented. Cyclosporine and antithymocyte immunoglobulin (ATG) are the treatments of choice.37, 42 Cases of simultaneous improvement of hepatitis features and pancytopenia after the initiation of immunosuppressive treatment imply that the exact immune-mediated mechanism can be responsible for both hepatic and bone marrow injury.37 Clinical data seem to confirm that, compared with the general population of patients with AA, patients with HAAA have similarly favorable results of bone marrow transplantation and immunosuppressive therapy.47
Interestingly, HAAA relatively often complicates liver transplantations performed due to hepatitis with fulminant organ failure, affecting 28% of patients in the pediatric population. The HAAA following liver transplantation can be diagnosed from 1 to 7 weeks after surgery. Earlier studies usually implemented a reduction of immunosuppression after the diagnosis of post-transplant HAAA to reduce the risk of severe infections.48 However, more recent data suggest very good results of the treatment with ATG in combination with cyclosporine A.49
Drug-induced AA can be caused by several pharmaceuticals used in hepatological practice, including interferon, azathioprine, propranolol, or spironolactone. Interferon treatment is known to cause immune-mediated side effects. Regarding the hematopoietic system, the most clinically significant phenomenon are cases of autoimmune hemolytic anemia. However, endogenous interferon is usually regarded as an important element of pathological pathways, leading to AA of other causes. Myelosuppression and AA have been also reported to complicate the administration of interferon in the treatment of chronic viral hepatitis.50, 51 Such reports are extremely rare, and with a declining indication for interferon use in hepatology, interferon-induced AA remains a marginal problem.
Summary
Anemia associated with liver disease is most frequently ascribed to blood loss from the gastrointestinal tract or micronutrient deficiency, with less common occurrence of hemolytic or aplastic anemia. However, both latter types of anemia require clinicians’ attention, as they can pose a significant threat to the patients if misdiagnosed and untreated. It is also important to remember that the diagnosis of one type of anemia does not preclude overlapping of other reasons for a decreased hemoglobin concentration. Therefore, a lack of improvement after the initial diagnosis and treatment of micronutrient deficiency or blood loss should trigger further diagnostics.
Hemolytic anemia is reported to complicate 1–14% of cases of advanced liver disease,1, 4 but in some clinical entities can be much more prevalent. Cases of immune-mediated hemolytic anemia can complicate autoimmune liver diseases, such as autoimmune hepatitis, PBC and PSC. Non-immune mediated hemolytic anemia in the context of liver disease can develop as acquired membranopathy of red blood cells. It can accompany virtually every severe liver disease. However, especially often, it can complicate alcohol liver disease. Non-immune mediated hemolysis can also be the first clinical manifestation of WD, particularly among younger patients. Non-immune mediated cases of hemolytic anemia caused by ribavirin are becoming less frequent due to a decreasing number of indications for the use of ribavirin in hepatology.
Aplasia occurring in the context of liver disease is a rare finding. However, cases associated with hepatitis are a well-recognized clinical entity. Viral agents most commonly trigger hepatitis-associated AA, but other noninfectious causes are also involved in the pathogenesis of AA.
Limitations
This review is burdened with several limitations that are typical for the methodology used. In comparison to systematic reviews, narrative reviews are regarded to have a more subjective character. However, authors have made every effort to avoid any bias in the process of paper selection. Moreover, this literature review was limited only to papers written in English, which could have affected its completeness. Since database search provided us with a very high number of potential matches to the topic of our review, we implemented strict rules of title and abstract selection. There is a risk that some articles rejected based on the title and abstract screening would provide valuable information if they were included.
As aplastic and hemolytic anemia are rarely diagnosed in liver disease, the available data are scarce and often of limited quality. The abovementioned clinical entities are frequently characterized based on series of case reports, and their treatment options are not tested in randomized clinical trials. Further research is needed to fully understand the prevalence of hemolytic and aplastic anemia in liver disease, and the peculiarities of their treatment.
Conclusions
Step-by-step approach to anemic patient with liver disease requires initially ruling out the most common underlying disorders. They include mainly blood loss and micronutrient deficiency. However, a considerable proportion of patients remain without a clear diagnosis even after that. In such cases, less common causes of anemia should be investigated, like hemolytic anemia and AA. Both are not homogenous clinical entities but umbrella terms for various diseases. Immune-mediated hemolytic anemia is known to complicate a number of autoimmune disorders, and is an uncommon finding among patients with AIH, PBC and PSC. Non-immune mediated hemolysis is usually regarded as more probable among patients with liver disease, and is often caused by acquired membranopathy of red blood cells. It should always be suspected in alcoholic liver disease and severe liver failure. When anemia complicates hepatitis of infectious origin, the evaluation toward AA may be indicated. Moreover, when hemolytic or AA is considered in differential diagnosis, the pharmacotherapy of a patient should be thoroughly analyzed, as many drugs have been associated with discussed hematological disorders.